Location: Home >> Detail
Immunometabolism. 2021;3(4):e210032. https://doi.org/10.20900/immunometab20210032

1 Biological Design Graduate Program, School for the Engineering of Matter, Transport, and Energy, Arizona State University, Tempe, AZ 85281, USA
2 Chemical Engineering, School for the Engineering of Matter, Transport, and Energy, Arizona State University, Tempe, AZ 85281, USA
3 Department of Immunobiology, University of Arizona, Tucson, AZ 85721, USA
4 Arizona Arthritis Center, College of Medicine, University of Arizona, Tucson, AZ 85721, USA
5 Materials Science and Engineering, School for the Engineering of Matter, Transport, and Energy, Arizona State University, Tempe, AZ 85281, USA
6 Center for Immunotherapy, Vaccines and Virotherapy, Arizona State University, Tempe, AZ 85281, USA
7 Biomedical Engineering, School of Biological and Health System Engineering, Arizona State University, Tempe, AZ 85281, USA
* Correspondence: Abhinav P. Acharya.
This article belongs to the Virtual Special Issue "Immunometabolism in Autoimmune Diseases"
An increasing number of findings highlight the fundamental role of metabolism in autoimmunity. This growing recognition of metabolic irregularities being a factor of immune dysregulation in autoimmunity has provided novel therapeutic opportunities. Although there have been advancements in the field of immunometabolism, it is important to note that strategies to engineer the immune system can also be utilized to modulate immunometabolism of specific immune cells that are involved in autoimmune rheumatoid arthritis (RA). Herein, we review the metabolic heterogeneity within RA, and discuss the potential therapies based on immunometabolism and immunoengineering concepts that may reinstate immune tolerance by targeting the metabolic irregularity within specific immune cells involved in the pathogenesis of RA.
Rheumatoid arthritis (RA) is a chronic, systemic autoimmune disorder marked by joint pain, synovial inflammation, and destruction of cartilage and bone in inflamed tissues [1,2] As a leading cause of disability, RA hinders the work ability and quality of life of millions of adults [3]. Although present therapies such as disease modifying anti-rheumatic drugs (DMARDs) can lower disease activity, there is currently no cure for RA, and its exact origin remains unknown [1,2]. Abnormal levels of autoantibodies, namely rheumatoid factor (RF) and anti-citrullinated protein antibodies (ACPA), often precede the onset of RA symptoms [4], and irregular immune regulation induces migration of T and B cells into the synovium. Because lymphocyte activation is crucial for RA pathogenesis, altering the metabolism of these immune cells presents a novel opportunity for RA treatment.
Understanding the importance of energy metabolism in immune cell function has allowed metabolic pathways to become a novel target for the treatment of autoimmunity [5,6]. Although energy metabolism is an essential chemical process that is utilized for cellular function, alteration of metabolism is an important regulator of immune cell activation and differentiation. Interestingly, this malfunction in energy metabolism has been linked to the development of autoimmune diseases such as systemic lupus erythematosus (SLE), multiple sclerosis (MS) and RA[5]. Specifically, in RA, reports have demonstrated an increase in glycolytic activity within autoimmune tissues [7]. For example, positron emission tomography/computed tomography (PET/CT) scans from RA patients have shown high levels of fluorodeoxyglucose (18F-FDG) uptake, a glucose analogue, in both inflamed joints and axillary lymph nodes [8].
Nonetheless, immune cell subsets within RA have unique alterations within their energy metabolism pathways, therefore, making RA a heterogenous autoimmune disease [9–11]. Recent progress in the field of immunometabolism has provided insight into the metabolic heterogeneity of RA and how metabolic processes can be modulated for the treatment of RA [5]. However, in order to overcome current clinical challenges in treating RA, therapeutics in developmental stages should consider incorporating strategies for targeted drug delivery to avoid non-specific immune modulation. Applying engineering principles to immunometabolism concepts can lead to the metabolic reprograming of specific immune cells that are implicated in the development and progression of autoimmunity. Herein, we review metabolic alterations within several immune cells implicated within the pathogenesis of RA, and discuss how an immunoengineering approach may reinstate immune tolerance by targeting the metabolism of specific immune cells.
The pathogenesis of RA is associated with several different immune cell types, including T cells, B cells, macrophages, dendritic cells (DCs), fibroblast-like synoviocytes (FLS), neutrophils, granulocytes and innate lymphoid cells (ILCs) [12]. Metabolic pathways play a significant role in the behavior of each of these cell types, and the specific pathways utilized by different cell types can be targeted to slow RA pathogenesis. For example, under normoxic conditions, a cell produces adenosine triphosphate (ATP) by converting glucose into pyruvate via the glycolytic pathway. Pyruvate is then converted into acetyl coenzyme A (acetyl CoA) to fuel the tricarboxylic acid (TCA) cycle for the production of nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2). This then feeds into the electron transport chain (ETC) for the synthesis of thirty-six ATP molecules [13]. Several immune cells with anti-inflammatory properties are known to rely on the TCA cycle and ETC to fulfill their cellular energy demand [14]. However, under hypoxic conditions, pyruvate is rerouted, and instead of entering the TCA cycle, it is converted into lactate for the quick generation of two ATP molecules [13]. Various immune cell types with pro-inflammatory properties rely on rapid ATP synthesis via glycolysis to meet their cellular energy demand [14–17]. The glycolysis pathway is also linked to the pentose phosphate pathway (PPP). Furthermore, the TCA metabolite, citrate, can initiate fatty acid synthesis (FAS) while fatty acid oxidation (FAO) generates acetyl CoA to further fuel the TCA cycle (Figure 1, All figures created using BioRender.com).
 Figure 1. Crucial metabolic pathways utilized to meet cellular energy requirements. Under normal oxygen conditions, a cell utilizes the glycolysis pathway to metabolize one glucose molecule into two pyruvate molecules. Pyruvate then enters the TCA cycle for the synthesis of thirty-six ATP molecules via the ETC. However, in the absence of oxygen, pyruvate is instead utilized to synthesize lactate for the quick generation of two ATP molecules.
Figure 1. Crucial metabolic pathways utilized to meet cellular energy requirements. Under normal oxygen conditions, a cell utilizes the glycolysis pathway to metabolize one glucose molecule into two pyruvate molecules. Pyruvate then enters the TCA cycle for the synthesis of thirty-six ATP molecules via the ETC. However, in the absence of oxygen, pyruvate is instead utilized to synthesize lactate for the quick generation of two ATP molecules.
In RA, several cell types, including macrophages [15] and DCs [18,19], predominantly rely on aerobic glycolysis upon activation, a phenomenon known as the Warburg effect [20]. DCs and macrophages can be found in inflamed joints and lymph nodes, which PET imaging has shown through increased 18F-FDG uptake in these areas in RA patients [21,22]. Therefore, glucose hypermetabolism in lymph nodes demonstrates the importance of secondary lymphoid organs in the pathogenesis of the disease [15,22]. Nonetheless, understanding the immunometabolism of not only the tissue that is affected but the associated lymphoid organs is paramount for developing effective therapies.
Cluster of Differentiation 4 (CD4+) T cells. In contrast to macrophages and DCs, T cells in RA have a low glycolytic flux, unlike healthy effector T cells, due to the downregulation of the glycolytic enzyme PFKFB3, which has been termed the anti-Warburg effect [23]. However, unsimilar to this finding, studies have also discovered increased glycolytic activity within T follicular helper (TFH) cells. TFH and T helper (TH)17 cells in RA mouse models [19,24]. Additionally, elevated levels of a glycolytic enzyme, hexokinase-2 (HK2), within lymphocytes infiltrating the joints of RA patients have been reported. Therefore, the glycolytic activity within T cells in RA has yet to be elucidated. Nonetheless, T cells play an important role in the induction and progression of RA.
Generally, when autoimmunity is not present, resting CD4+ T cells utilize oxidative phosphorylation (OXPHOS) and break down fatty acids for generating energy, while activated CD4+ T cells, which have a high need for nutrients, conduct aerobic glycolysis. However, CD4+ T cells in patients with RA may downregulate PFKFB3, an allosteric enzyme that catalyzes an early step in glycolysis and plays an important role in CD4+ T cell activation. As a result, CD4+ T cells in RA patients can have a low glycolytic flux, ATP, lactate and pyruvate, in turn reducing mitochondrial metabolism [15,23]. Nevertheless, unlike other cell types, CD4+ T cells may have the unique ability to proliferate and secrete cytokines even when energy-deprived [25], and consequently, CD4+ T cells in RA can hyper-proliferate despite their downregulation of PFKFB3 [15]. However, as previously mentioned, this finding of RA CD4+ T cells displaying low glycolytic flux is still highly controversial. Studies have claimed that TFH, TH1, TH17 cells in RA rely on glycolysis, similar to activated healthy T cells [24,26,27]. However, T cells within RA modify the glycolysis pathway to result in decreased ATP production and increased production of molecular precursors required for rapid proliferation. This in turn leads to a reduction of reactive oxygen species (ROS) and an ultimate irregularity in cellular behavior, resulting in increased proliferation and differentiation of inflammatory T cell subsets [28].
However, in addition to impaired glycolysis, RA CD4+ T cells can have upregulated levels of glucose-6-phosphate dehydrogenase (G6PD), which shunts glucose into the PPP. Utilizing the PPP increases levels of NADPH, which converts glutathione disulfide to its reduced form glutathione [23]. Glutathione, an antioxidant, is responsible for scavenging ROS, which is a chemically reactive species that can cause cell senescence and tissue damage [15,29]. In healthy cells, NADPH oxidase (NOX)-dependent ROS production balances ROS scavengers, so the excess production of glutathione in CD4+ T cells with RA can causes low cellular ROS. In fact, lower levels of ROS production in CD4+ T-cells due to NOX2 deficiency have been shown to cause an increased risk for arthritis and more severe inflammation [23,30]. Although increased ROS has been associated with many diseases, such as cardiovascular diseases, due to its ability to damage cellular lipids, proteins, and deoxyribonucleic acid (DNA), low levels of ROS is also harmful due to the suppression of redox-sensitive signaling which can be utilized for effective intracellular communication [23]. The scavenging of ROS caused by the PPP impairs redox-signaling, which can lead to the hyperproliferation of CD4+ T cells. Low levels of ROS can inhibit the activation of the protein kinase Ataxia telangiectasia mutated (ATM), which is redox-sensitive and regulates the process of the cell cycle in dividing cells. The diminished activation of ATM may allow RA CD4+ T cells to bypass the G2/M cell cycle checkpoint and hyper-proliferate, causing premature senescence, and drives T-cell differentiation into pro-inflammatory TH1 and TH17 lineages. Consequently, artificially increasing the levels of ROS using menadione (an analog of 1,4-naphthoquinone) or buthionine sulphoximine (BSO) (limits tissue glutathione concentration), downregulates pro-inflammatory transcription factors and cytokines, decreasing T cell hyperproliferation and synovial inflammation [7].
However, different autoimmune diseases have a unique pattern of metabolic changes that occur during pathogenesis and disease progression [23]. For example, T cell metabolism also plays an important role in other autoimmune diseases, such as SLE and experimental autoimmune encephalomyelitis (EAE), which is the mouse model for human MS. In contrast to T cells from RA patients, in EAE, the metabolism of T cells is similar to that of normal healthy effector T cells, with a reliance on aerobic glycolysis for energy needs. Increased expression of glycolytic enzymes was found to fuel aerobic glycolysis in EAE T cells and promote the production of pro-inflammatory interferon γ (IFN-γ) and interleukin 2 (IL-2) cytokines [31]. Similarly, T cells in progressive MS patients upregulate glycolysis and are characterized by higher lactate levels[32]. However, similarities within T cell metabolism between RA and SLE have been reported. Similar to RA, T cells from SLE patients predominately rely on mitochondrial oxidation for their energy needs, however, SLE T cells have increased ROS production [33]. Additionally, SLE T cells contain defects in lipid metabolism and membrane raft formation which leads to an alteration in T-cell receptor (TCR) signaling and causes abnormal T cell activation [23]. Interestingly, both in RA and SLE, as well as in a number of other autoimmune diseases, the activation of these abnormal T cells leads to the generation of autoantibodies via activation of B cells. In SLE specifically, these changes in adaptive immune cell activity (B and T cells) can be traced back to the effects of lupus susceptibility genes (Sle1-Sle3). A study crossing NZM2410 and C57BL/6j mice found that Sle1 and Sle2 contribute to aberrant B cell activation by stimulating the production of autoantibodies and lowering the activation threshold of B cells. Moreover, Sle3 promotes T cell dysregulation and reduces activation-induced cell death in CD4+ T cells, in turn prompting irregular T cell activation [34]. It will be interesting to study if some of these irregular activities are indeed caused by the increased ROS production. Nonetheless, in RA, SLE, EAE, and MS changes in T cell metabolism heavily influence the development of chronic autoimmunity and inflammation.
Targeting the alterations in CD4+ T cell metabolism during autoimmune disorders offers a unique method to treat these diseases. The use of the PPP and the subsequent lack of ROS in RA CD4+ T cells can be responsible for T cell hyperproliferation, as the impairment of redox-sensitive signaling may allow CD4+ T cells in RA to bypass cell checkpoints. Consequently, replenishing the levels of ROS has shown to inhibit this hyperproliferation and reduce synovial inflammation, making an increase in ROS a potential therapy for RA [7]. In contrast to RA, in SLE, the pathological reliance on mitochondrial metabolism in T cells is consistent with the discovery that SLE is suppressed by the inhibition of mitochondrial F1/F0-ATPase by the immunomodulatory benzodiazepine Bz-423 [33,35]. Moreover, since T cell metabolism in EAE is dependent on glycolysis, a delivery of the glycolytic inhibitor 2-deoxyglucose (2-DG) was found to lower production of the pro-inflammatory cytokine IL-17 and reduce T cell proliferation [33,36]. Although there is a variance in which bioenergetic pathways are utilized within T cells of RA, SLE and MS patients, T cell metabolism does present a potential target to treat and prevent autoimmunity.
TFH cells. A subset of CD4+ T cells that express CXC-chemokine receptor 5 (CXCR5), play an important role in the pathogenesis of RA and the development of chronic autoimmunity[37]. TFH cells are known to regulate B cell activation, promote antibody affinity maturation, and initiate the germinal center (GC) reaction. In RA, autoreactive TFH cells are essential for the production of autoantibodies by B cells and the formation of synovial ectopic lymphoid structures (ELS) (aggregates of B and T cells in chronically inflamed tissues that resemble GCs) [37,38]. Additionally, levels of circulating TFH (c TFH) cells and activated B cells have been shown to increase with the onset of RA and are correlated with disease severity, suggesting that these cells may be used as biomarkers for RA[39].
Although the joint is the main site of inflammation and metabolic changes, emerging studies have found a correlation between gut metabolism and RA [40]. The gut microbiome, which is the collection of microbes in the intestines, has been shown to influence RA pathogenesis by accelerating the production of pro-inflammatory mediators [40–42]. A study using the K/BxN arthritis model found that gut microbiota such as segmented filamentous bacteria (SFB) induce TFH differentiation in Peyer’s patches (PP) via DC-mediated inhibition of the IL-2 signaling pathway [40]. These PP TFH cells later migrate into systemic sites to promote autoantibody production there. An additional study utilizing a K/BxN mouse model found that autoreactive TFH cells are highly dependent on glycolysis, and the inhibition of glycolysis through 2-DG significantly reduced levels of autoreactive TFH cells and alleviated synovial inflammation[24]. In contrast, glycolytic inhibition does not affect levels of pathogen-specific TFH cells, indicating that non-autoreactive TFH cells do not rely on glycolysis for energy in K/BxN mice. Consequently, lowering glycolytic flux may be useful for treating RA by reducing levels of autoreactive TFH cells while maintaining immunity against pathogens [43,44].
TH17 and TH1 cells. While TFH cells contribute to RA pathogenesis by influencing B cells, TH17 cells directly contribute to synovial inflammation and bone erosion through their production of pro-inflammatory cytokines, including IL-17, TNF-α, IFN-γ, and granulocyte-macrophage colony-stimulating factor (GM-CSF) [9]. Gut microbiota have been shown to influence the progression of RA via TH17 cells. K/BxN mice housed under germ-free (GF) conditions has been shown to have decrease TH17 levels and the introduction of SFB was shown to accelerate the pathogenesis of RA by promoting TH17 levels [42]. In terms of metabolic pathways, TH17 cells have shown to upregulate glycolytic flux through hypoxia-inducible factor 1-alpha (HIF-1α) induction and mechanistic target of rapamycin (mTOR) signaling [45]. Notably, HIF-1α deficiency has been shown to reduce the expression of glycolytic molecules and therefore limit TH17 development [9,36]. Additionally, de novo FAS is essential for TH17 cell differentiation, and therefore, inhibiting acetyl-CoA carboxylase 1 (ACC1), an enzyme required for de novo FAS, with the ACC-specific inhibitor soraphen A (SorA) has been shown to limit TH17 production[9,45,46]. Consequently, targeting the mTOR/HIF-1α pathway and activating adenosine monophosphate-activated protein kinase (AMPK), which is an inhibitor of de novo FAS, are both possible therapies for TH17-mediated RA symptoms [45]. Similar to TH17 cells, TH1 cells are known to be highly glycolytic with increased expression of the glucose transporter 1 (GLUT1) [47,48]. Interestingly, TH1 cells have shown to have an increased frequency within the joint of RA patients [27] and have also shown to heavily rely on glycolysis or glutamine pathways for cellular function [47]. A recent study by Pucino et al. found that lactate buildup in the synovium due to higher rates of aerobic glycolysis promotes pro-inflammatory metabolism in CD4+ T cells. High levels of extracellular lactate upregulate the lactate transporter SLC5A12, which is expressed by CD4+ T cells, and contribute to increased IL-17 production and fatty acid synthesis [49]. Furthermore, in vitro, studies reported that L-type amino acid transporters, which influence the uptake of phenylalanine, tyrosine, leucine, arginine, and tryptophan, are necessary for TH1 and TH17 expansion and differentiation [47,50]. However, there is currently limited research regarding the metabolism of TH1 cells in RA in patients specifically.
Regulatory T (TREG) cells. Another type of T cell that can be targeted in RA are TREGs, which can prevent autoimmunity by suppressing the function of autoreactive lymphocytes [51,52]. Several studies have found elevated levels of TREGs in RA SF, but studies have found conflicting results regarding the levels of TREGs in peripheral blood of RA patients [43]. Similarly, it is unclear whether RA affects the ability for TREGs to suppress effector T cell proliferation [51]. One study found that TREGs in RA are unable to block the secretion of pro-inflammatory cytokines by effector T cells, although anti-TNFα therapy restored normal functions [53]. In contrast, another study has found that TREGs in RA retain their suppressive functions and proposed that synovial inflammation occurs because CD4+Foxp3−CD25− T cells in RA are activated to an extent that their hyperproliferation cannot be efficiently suppressed by TREGs [54]. Consequently, further research is necessary to clarify the role of TREGs in RA pathogenesis. TREG metabolism has been shown to influence cell proliferation and function, and the metabolism of these cells alters due to a balance between toll-like receptor (TLR) signaling and the transcription factor Foxp3 [55]. Upon activation by TLR signals that promote proliferation, TREGs mainly utilize aerobic glycolysis, as TLR-induced phosphoinositide 3‐kinase (PI3K)-Akt-mTORC1 signaling increases expression of the glucose transporter GLUT1 and thus increases glycolytic flux. However, increased GLUT1 expression has been shown to accompany decreased suppressive capacity of TREGs by downregulating Foxp3 expression [55]. Suppressive TREGs expressing high levels of Foxp3 rely more on oxidative and catabolic metabolism because Foxp3 decreases PI3K-Akt-mTORC1 signaling and therefore lowers glycolytic flux[55]. Consequently, inhibiting glycolysis in the rheumatoid synovium may be a possible therapy for RA, as glycolysis is essential for most effector cells but not for TREG suppressive functions [56].
CD8+ T cells. Although CD8+ T cells are not as widely studied as CD4+ T cells in the context of RA, CD8+ T cells have been shown to play an important role in RA progression [57]. CD8+ T cells differentiate into heterogeneous cell subtypes that differ based on their expression of surface and intracellular markers and their production of cytokines. In RA, differentiation is skewed towards inflammatory CD8+ phenotypes, and levels of CD8+ T cells have been shown to correlate with disease severity, making these cells possible biomarkers for RA progression [57,58]. CD8+ T cells are known to switch from a reliance on OXPHOS to aerobic glycolysis, PPP, and glutaminolysis upon activation, and inhibiting glycolysis using 2-DG has been shown to limit effector CD8+ functions [59,60]. Consequently, blocking glycolysis in CD8+ T cells may help alleviate synovial inflammation in RA patients. Additionally, IL-17-producing CD8+ T (cytotoxic T cells—TC17) cells have been shown to play a possible role in RA pathogenesis. TC17 cells were found at elevated levels in the peripheral blood of RA patients, specifically in those with high disease activity [61,62]. However, little is known about the metabolism of TC17 cells in RA, indicating a need for further research.
B-cells. In addition to T cells, B cells play a significant role in the pathogenesis of RA. Autoreactive B cells produce autoantibodies, namely ACPAs and RFs. ACPAs, which identify citrulline-modified peptides and proteins, and RFs, which are specific for the Fc portion of Immunoglobin G (IgG), both serve as diagnostic markers in RA [12]. Although increasing RF levels alone in healthy individuals does not induce synovial inflammation, RFs aggravate inflammatory processes in immune complexes[63]. Moreover, citrullination, the post-translational replacement of arginine with citrulline, may be caused by abnormal protein metabolism at the sites of irregular apoptosis, and citrullination has been suggested to predispose individuals to RA [64]. ACPAs increase bone erosion in the joints of RA patients and stimulate immune effector functions, including the activation of Fcγ receptors [65]. In addition to the production of autoantibodies, B cells also advance RA pathogenesis by secreting pro-inflammatory cytokines, including tumor necrosis factor-alpha (TNF-α) and IL-17 [11,12]. An in vivo study found higher glycolytic flux and lactate production in B cells upon activation by lipopolysaccharide (LPS), indicating the utilization of aerobic glycolysis. Increased levels of glycolysis were shown to be essential for B cell function, and the impairment of glycolysis through dichloroacetate (DCA), an inhibitor of pyruvate dehydrogenase kinase (PDHK), decreased B cell proliferation and antibody production [66]. Consequently, it is likely that activated RA B cells rely on glycolysis for energy and autoantibody production, and therefore, targeting glycolysis in B cells provides a novel method to halt the production of autoantibodies and the progression of RA [67]. However, recent literature provides contradictory results in demonstrating that B cells within the GC have diminished glycolytic activity and have increased OXPHOS and oxidize fatty acids for the expansion of B cells within the GC and for the high production of class switching antibodies [68,69]. Therefore, targeting OXPHOS within the GC may alter the function of GC B cells.
Nonetheless autoreactive B cells migrate into the synovium and promote inflammation. On the other hand, IL-10-producing regulatory B (Breg) cells, which suppress inflammation, are present at a lower frequency in RA patients. In fact, because of their immunosuppressive properties, increasing the number or frequency of Breg cells in inflamed tissues could reduce disease activity. A recent study showed that supplementing butyrate, a microbiota-derived short-chain fatty acid, in RA patients can lower disease severity by supporting Breg function and inhibiting germinal center (GC) B cell and plasmablast differentiation [70].
Macrophages. Macrophages play an important role in innate immunity and are essential for the pathogenesis of RA. Macrophages secrete pro-inflammatory mediators, and also assist in the recruitment of lymphocytes by acting as antigen-presenting cells (APCs) [12]. While RA T cells in the synovium milieu are energy-deprived due to their low production of ATP, macrophages within the synovium are characterized by a hyper-metabolic state, where they have increased glycolytic flux, in turn leading to decreased glucose and increased lactate in the extracellular space [15]. Interestingly, macrophages in RA tissue upregulate the glucose transporters GLUT1 and GLUT3 along with the glycolytic enzymes enzyme pyruvate kinase 2 (PKM2), PFKFB3 and HK2 [15]. In fact, even resting macrophages in RA pathology have been found to be in a hyper-metabolic state. RA macrophages have shown to consume excess glucose for ATP production when in a resting state. This increase in glucose uptake causes an exponential increase in mitochondrial oxidation, resulting in the production of ROS by the ETC. Consequently, RA macrophages have high levels of ROS (unlike RA T cells), activating the redox-sensitive glycolytic protein, PKM2 [15]. PKM2, which acts as a protein kinase, phosphorylates the transcription factor signal transducer and activator of transcription 3 (STAT3), stimulating the production of pro-inflammatory IL-1β and IL-6 cytokines [15]. Macrophages are known to be major contributors to the cytokine-rich synovial milieu, and their production of pro-inflammatory cytokines is closely related to their high glycolytic flux [15]. Additionally, succinate plays a significant role in the immune response of macrophages, and a metabolic profiling study has found elevated levels of succinate in RA SF [71]. In in vitro conditions, macrophages stimulated with LPS accumulate and release succinate, possibly due to a “broken” Krebs cycle [also known as TCA cycle [72], and these high levels of extracellular succinate have been shown to stabilize HIF‐1α and increase IL-1β production [73,74]. Similarly, an in vivo study has demonstrated the secretion of succinate by macrophages in RA upon activation by TLR ligands. Moreover, RA macrophages sense extracellular succinate via G-protein coupled receptor 91 (GPR91)/succinate receptor 1 (SUCNR1), which stimulates IL-1β production and drives synovial inflammation. As a result, GPR91 antagonists may be a possible treatment for RA and have already been shown to inhibit IL-1β release [73]. In addition to macrophages, other antigen presenting cells (APCs) also play an important role in the pathogenesis of RA.
DCs. DCs are APCs linking adaptive and innate immunity, and therefore, are crucial for the activation of T and B cells in RA pathogenesis. While healthy resting DCs with anti-inflammatory phenotypes utilize mitochondrial oxidation, DCs switch to a pro-inflammatory phenotype and rely on aerobic glycolysis upon activation [16]. This increased aerobic glycolysis via glycolytic enzyme HK2, which is activated by the kinases TANK-binding kinase 1 (TBK1), IKKε, and protein kinase B (PKB), induces the synthesis of fatty acids and other anabolic demands immediately upon DC activation. Consequently, inhibition of HK2, using 2-DG, has been shown to limit DC activation by impairing glycolysis [13,75]. The HK2 inhibitor, 2-DG, is a non-metabolizable glucose counterpart that is capable of blocking glucose phosphorylation and impeding protein glycosylation [76]. Increased reliance on glycolysis in RA DCs has been further confirmed by a recent finding that3-bromopyruvate (BrPA), another HK2 inhibitor, decreased the activation of DCs in vitro and significantly lowered arthritis scores in SKG mice [19]. In later stages after activation, the induction of inducible nitric oxide synthase (iNOS) by mammalian target of rapamycin (mTOR) and the stabilization of HIF‐1α solidify the reliance upon glycolysis, as iNOS‐derived nitric oxide (NO) suppresses the ETC [13]. It is important to note that there are several known glycolytic targets, such as GLUT1, PFKFB3 and lactate dehydrogenase (LDH), which have all shown to successfully inhibit the glycolytic pathway [77]. Additionally, DCs in the synovium are able to sense and adapt to surrounding metabolites, including succinate, butyrate, and ATP [13]. In fact, the succinate receptor, GPR91, which is highly expressed by DCs [78], serves as a chemotactic, guiding DCs into lymph nodes and mediating the expansion of pro-inflammatory TH17 cells in lymph nodes. Therefore, a blockade of GPR91 is further shown to be a possible therapy for RA by reducing DC proliferation and decreasing the frequency of TH17 cells in lymph nodes [13,79]. Additionally, DCs are known to release glutathione, which is cleaved into cysteine, lowering extracellular redox potential and enhancing effector T cell proliferation [80,81]. This process may cause further T cell-mediated tissue damage in RA.
Neutrophils. As members of the phagocytic innate immune system, neutrophils are essential in the initial stages of RA pathogenesis. Neutrophils in RA secrete ROS and reactive nitrogen species (RNS), which are produced by NOX2 and iNOS-derived NO respectively, causing damage to articular cartilage and bone [82]. However, the impact of ROS and RNS on inflammation-mediated progression of RA is not clear. A study using the K/BxN serum transfer model of RA found that the synovial inflammation in iNOS2 knockout mice and gp91phox (NOX2)-deficient mice was just as severe as controls, suggesting that ROS and RNS do not play a role in innate immunity-mediated inflammation [83]. In addition to ROS and RNS, neutrophils in the synovium also produce granules of degradative enzymes, pro-inflammatory cytokines, and neutrophil extracellular traps (NETs—web-like structures of chromatin and granule-derived peptides and enzymes) [84]. NETs can contain citrullinated autoantigens, which stimulate the production of ACPA by B cells and contribute to the development of autoimmunity [84]. Additionally, the production of NETs and other neutrophil effector functions predominantly rely on glycolysis and the PPP for energy, as neutrophils generally contain few mitochondria [85,86]. Further, an in vitro metabolic profiling study on phorbol 12-myristate 13-acetate (PMA)-activated neutrophils found that upon activation, levels of NADPH and lactate increased, while levels of glucose initially increased but later decreased. The increase in NADPH suggests that activated neutrophils rely on the PPP for energy; therefore, blocking this pathway in neutrophils during early stages of inflammation may slow RA pathogenesis [87].
FLS. FLSs are the most common cell type in the rheumatoid pannus (a type of abnormal growth that consists of fibrovascular and granulation tissue), and they greatly contribute to joint destruction and synovial hyperplasia in RA. While healthy FLSs help protect and lubricate the synovial lining, FLSs in RA acquire an aggressive phenotype that accelerates joint damage. FLSs in RA are characterized by the production of inflammatory cytokines and matrix metalloproteinases (MMPs), migration and invasion into bone and cartilage, and resistance to apoptosis [81,88]. Consequently, this aggressive phenotype puts FLSs into a hyper-metabolic state with increased glycolysis, amino acid metabolism, and protein biosynthesis [13,89]. Additionally, synovial cells and tissues (e.g., synovial membrane/endothelium, synovial fibroblasts, naïve and effector T cells, monocytes, DCs and various subsets of macrophages) within the nutrient depleted and hypoxic synovium adapt and alter key molecular switches (e.g., PFKFB3, mTOR, PKM2, G6PD, AKT and iNOS) within bioenergetic pathways for survival and expansion [23]. Furthermore, the hypoxic conditions and high expression of HIF‐1α in the synovium promote an invasive behavior in FLSs, such as an upregulation in glycolytic proteins regulated by HIF‐1α, including the GLUT1, HK2, and LDH [88]. Consequently, previous studies have shown that impairing glycolysis through HK2 and PFKFB3 inhibitors, as well as the glycolytic inhibitors 2-DG and 3-bromopyruvate (BrPa), can lower the activity of FLSs and mitigate joint damage [13,90]. Furthermore, the uptake of glutamine can lead to the activation of mTOR, independent of glutaminolyisis, to promote the invasive behavior of FLSs [91]. Interestingly, it has also been observed that RA FLSs rely on glutamine metabolism for energy, and inhibiting glutamine was found to reduce FLS proliferation [92].
γδ T cells, basophils, eosinophils and mast cells. Gamma-delta (γδ) T cells, which are divided into Vδ1 and Vδ2 T cells, contribute to the inflammatory synovial milieu in RA through the production of pro-inflammatory cytokines. Vδ2 T cells specifically, which secrete IFN-γ, TNF-α, and IL-17, have been found to migrate to joints and accumulate in RA SF, possibly due to the expression of chemokines CCR5 and CXCR3 [93]. However, another study has found that synovial γδ T cells only produce IL-17 in collagen-induced arthritis (CIA) but not in human RA, so further investigation is warranted [94]. In addition to γδ T cells, basophils and mast cells, which are granulocytes that release degrading compounds such as histamine, also further RA pathogenesis upon activation by ACPA, TLR ligands, and other stimuli [95,96]. Mast cells’ effector functions have been shown to rely on both glycolysis and OXPHOS, and glycolytic inhibitors, such as 2-DG and DCA, as well as rotenone, which impairs mitochondrial metabolism, reduced the ability of mast cells to produce cytokines and degranulate [86,97]. Consequently, targeting both glycolysis and OXPHOS in synovial mast cells may be possible therapies for RA. Another cell type that influences RA pathogenesis is eosinophils, which can indicate a poor prognosis when abundant in RA SF. High levels of eosinophils, also known as eosinophilia, are correlated with poor responses to RA treatments, namely DMARD therapy, and indicate higher disease severity [98]. Interestingly, increased eosinophil and TH2 responses, which can occur from a N. brasiliensis infection, have been shown to reduce inflammation and bone erosion in both the serum-induced arthritis (SIA) model and human TNF transgenic (hTNFtg) mice. Further, eosinophil-deficient ΔdblGATA mice were found to have higher severity of arthritis, and the reintroduction of eosinophils alleviated inflammation and initiated the resolution of symptoms [99]. A metabolic profiling study of eosinophils has shown a high oxygen consumption rate (OCR) in eosinophils compared to neutrophils and similar extracellular acidification rates (ECAR) between eosinophils and neutrophils, indicating that eosinophils may also rely on both glycolysis and mitochondrial oxidation for energy [100]. It will be interesting to learn whether the anti-inflammation effect of eosinophil relies on glycolysis or OXPHOS.
ILCs. As the innate counterparts of T cells, innate lymphoid cells (ILCs) play important roles in both the progression and resolution of RA [101,102]. Recent studies have classified ILCs into five subsets based on development and function: natural killer (NK) cells, ILC1s, ILC2s, ILC3s, and lymphoid tissue-inducer (LTi) cells [102,103]. ILC2s are known to promote anti-inflammatory responses, whereas ILC1s and ILC3s are associated with the induction of inflammatory responses. IL-9 stimulates the expansion of ILC2 and this subsequently leads to the activation of TREGs, TH2 cells and M2 macrophages for an ultimate suppression of TH17 cells within the synovium. As opposed to ILC2s, ILC1s and ILC3s stimulate pro-inflammatory responses within the synovium by activating TH1 and TH17 cells, for the induction of M1 macrophages [104]. A study investigating the frequencies of ILC subsets in lymph node (LN) biopsy specimens from patients with early RA and “at-risk” patients (positive for RF and/or ACPA but lacking clinical signs of RA) found a shift towards inflammatory phenotypes [104]. RA patients were found to have elevated levels of ILC1s and ILC3s, which produce pro-inflammatory cytokines, and lower levels of LTi cells, which may indicate impaired LN remodeling and an autoimmune‐prone LN microenvironment [104]. Further, CD3–CD56bright NK cells were found to be at elevated levels in RA SF compared to peripheral blood [105]. A study using the K/BxN SIA model of arthritis found that the production of IL-9 by ILC2s plays an important role in the resolution of RA, and treatment with IL-9 induced ILC2-dependent TREG activation and accelerated resolution of arthritis [106]. Moreover, ILC2s were found to be reduced in the peripheral blood of RA patients compared to patients in remission, and the level of ILC2s negatively correlated with disease activity [106].
Metabolic pathways have been shown to play an important role in ILC function; however, there is limited research on ILC metabolism specifically in RA. Interestingly, the enzyme arginase-1 (Arg1), which metabolizes the amino acid l-arginine to create urea and ornithine, is essential for ILC2 proliferation and cytokine production in type 2 inflammation. Contrary to the role of ILC2’s in RA, the reduction of ILC2s, by inhibiting Arg1, was found to decrease ILC2-mediated airway inflammation in lung disease by disrupting amino acid metabolism, and reducing ILC2 glycolytic capacity [107]. This suggests that the role of ILC2s may vary in different inflammatory diseases. However, it is important to note that ILC2s have been shown to rely on different metabolic pathways depending on the inflammatory stimulus and tissue environment. While lung ILC2s that promote airway inflammation mainly utilize glycolysis and OXPHOS, intestinal ILC2s rely more on FAO for anti-helminth immunity [108]. Interestingly, the intestinal immune system, such as gut-residing TH17 cells, DCs of Peyer's patches, has shown to be implicated in RA pathogenesis [42,108–110]. Therefore, the finding of intestinal ILC2 metabolism in helminth immunity may provide some insight into ILC2 metabolism in RA and its impact on additional intestinal immune cells. However, there has been little research investigating the metabolism of ILC2s in RA specifically. Moreover, little is known about the metabolism of ILC1s and ILC3s, indicating that further studies are required.
NK cells: NK cells have similar metabolic activity to healthy T cells, which is consistent with the common lymphoid lineage and shared effector functions between NK and T cells [111]. Mature naïve NK cells are metabolically quiescent and rely on OXPHOS and FAO, while activated NK cells have increased metabolic activity, with elevated levels of both glycolysis and OXPHOS [111–113]. However, activated NK cells upregulate glycolysis at higher rates than OXPHOS, indicating a shift towards glycolysis upon activation [111,114]. Mammalian target of rapamycin complex 1 (mTORC1), which regulates both immune responses and metabolism, has been shown to mediate metabolic reprogramming of NK cells upon activation, which is essential for NK effector functions. An in vivo study found that mTORC1 activity is essential for IFN-γ and granzyme B production by NK cells, and mTORC1 signaling was found to increase metabolic activity and glucose uptake upon activation [114]. Consequently, inhibiting mTORC1 via rapamycin in NK cells impairs glycolysis by limiting the glucose transporter GLUT1 and glycolytic enzymes HK2 and LDHA, and the inhibition of mTORC1 has also been shown to disrupt NK cell cytotoxicity [114,115].
The inflammatory nature of each of these immune cells may influences the hypoxic environment within the synovium, in turn contributing to RA pathogenesis by influencing the metabolism of synovial cells. Hypoxia stimulates increased expression of HIF‐1α, an important transcription factor that induces mitochondrial dysfunction and increased glycolysis in synovial cells [116]. HIF‐1α interacts with nuclear factor kappa light‐chain‐enhancer of activated B cells (NF‐κB), Notch‐1 intracellular domain (Notch‐1), Janus kinase/signal transducers and activators of transcription (JAK-STAT), and the PI3K-PKB/Akt pathway to produce inflammatory conditions [13]. Additionally, HIF‐1α stimulates glycolysis via the PI3K-PKB/Akt pathway, which inhibits glycogen synthesis by limiting glycogen synthase kinase 3β (GSK-3β) [117]. Importantly, levels of hypoxia are strongly correlated with synovial inflammation and production of pro-inflammatory cytokines, including TNF-α, IL-1β, and IFN-γ. Additionally, hypoxic conditions in the synovium have been shown to prompt angiogenesis, migration of inflammatory cells into the synovium, increased cell survival pathways, and increased MMP-mediated tissue degradation [118]. Notably, the Krebs cycle metabolite, succinate, is abundantly present in RA SF. Succinate has been shown to promote abnormal angiogenesis through HIF-1α induction [88], which in turn further promotes inflammation by facilitating immune cell recruitment (Figure 2) [13]. Therefore, RA therapeutics, that focus on immunometabolism, can prove to be an effective treatment option.
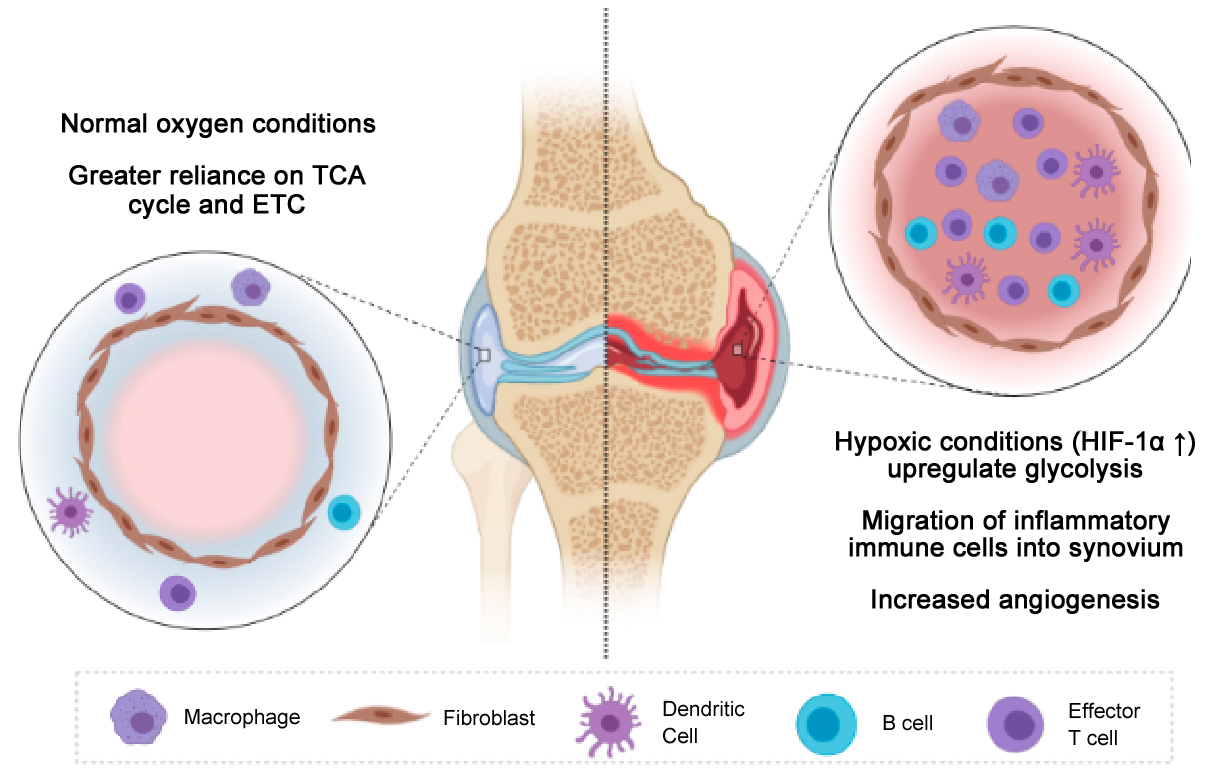 Figure 2. Normal versus hypoxic conditions within the synovium. The hypoxic environment within the RA synovium leads to an increase in the migration of inflammatory immune cells within the synovium, where synovial cells adapt to the nutrient deprived and hypoxic environment by increasing angiogenesis and upregulating glycolysis to meet the metabolic demand required for the inflammatory cells to survive and expand within the synovium.
Figure 2. Normal versus hypoxic conditions within the synovium. The hypoxic environment within the RA synovium leads to an increase in the migration of inflammatory immune cells within the synovium, where synovial cells adapt to the nutrient deprived and hypoxic environment by increasing angiogenesis and upregulating glycolysis to meet the metabolic demand required for the inflammatory cells to survive and expand within the synovium.
Currently, treatments for RA prioritize the achievement of remission or low disease activity through the treat-to-target (T2T) strategy, which emphasizes the constant monitoring of disease activity and subsequent adjustment of medication to maintain a certain target [119]. The T2T approach has shown to be particularly effective for those with early RA. Early treatment with DMARDs has shown to be effective in short-term disease management, and may improve long-term outcomes for RA patients who initiate DMARD therapy promptly after RA onset [119,120]. However, some have argued that the T2T strategy worsens long-term outcomes for certain patients due to the prioritization of a target over the drug being administered, resulting in a lack of personalized treatment. For example, common treatments for RA, such as biological DMARDs (bDMARDs), are not effective for all patients and can have long-term side effects, including tuberculosis infection, and liver and medullary toxicity [121].
Immediately upon diagnosis with RA, the conventional synthetic DMARD (csDMARD) methotrexate (MTX) is usually administered, either alone as a monotherapy or in combination with other csDMARDs and either with or without glucocorticoids (GCs) [122,123]. Glucocorticoids prevent the chemotaxis and access of leukocytes to sites of inflammation and also impede immune cell function while suppressing humoral responses with inflammatory properties [124]. Furthermore, glucocorticoids trigger gluconeogenesis in the liver, restrain glucose uptake in muscle and adipose tissue, and incite the breakdown of fat in adipose tissue, therefore playing a role in fat and glucose metabolism [125,126]. Whereas, MTX is a folic acid antagonist and has become the standard treatment for RA and one of the most popular drugs worldwide for this disease [127,128]. As a folate analogue, MTX inhibits dihydrofolate reductase (DHFR), in turn preventing folate reduction, which leads to an increase in the anti-inflammatory agent adenosine and a decrease in purine and pyrimidine biosynthesis, in turn blocking the production of DNA and RNA and therefore limiting cell proliferation (Figure 3) [129,130]. Nayak et al. illustrate how MTX can modulate the gut microbiota by targeting the conserved purine and pyrimidine metabolic pathways [131]. Interestingly, Bacteroides were the most impacted phyla when introduced to MTX in culture. Additionally, the colonization of germ free mice colonized with the post-MTX stool samples from RA patients demonstrated a decrease in activated T cells, TH17 cells and myeloid cells in the spleen and a decrease in activated T cells, TH17 cells and myeloid cells in the intestinal mucosa in the presence of inflammation. Therfore, further investigations are warranted to understand how DMARDs may influence the bacterial and cellular populations within the gut. In addition to targeting purine and pyrimidine biosynthesis [132], current research suggests that there are several other mechanisms of action through which MTX reduces the symptoms of RA, including ROS generation, polyamine inhibition, a reduction in chemotaxis and adhesion of inflammatory cells, and alteration of cytokine production [133,134]. Nonetheless, adenosine signaling is the most accepted explanation as the mechanism of action in RA [133], suggesting, that in RA, MTX may function more as an anti-inflammatory rather than antiproliferative drug [129,133]. Furthermore, increased adenosine release due to MTX has shown to limit the adhesion of neutrophils to connective tissue cells, thus limiting the infiltration of connective tissue cells by neutrophils [135]. Overall, there are various ways that MTX can exert anti-inflammatory effects directly or indirectly on cells involved in RA pathogenesis. For example, MTX can modulate inflammation by reducing monocyte populations via apoptosis, altering enzyme, cytokine, and gene expression, and promoting neutrophil chemotaxis. Furthermore, MTX increases levels of cyclo-oxygenase-2 (COX-2) production IL-1 receptor antagonist (IL1ra), soluble TNFα receptor (sTNFR), and transforming growth factor beta 1 (TGF1-β [129].
 Figure 3. Mechanisms of MTX action. Inhibition of DHFR and an increase in adenosine signaling are two well established mechanisms of action of MTX. The inhibition of DHFR prevents cell proliferation and the increase in adenosine signaling leads to the inhibition of various pro-inflammatory pathways.
Figure 3. Mechanisms of MTX action. Inhibition of DHFR and an increase in adenosine signaling are two well established mechanisms of action of MTX. The inhibition of DHFR prevents cell proliferation and the increase in adenosine signaling leads to the inhibition of various pro-inflammatory pathways.
MTX has proven to be effective in reducing inflammatory RA symptoms and has shown to outperform other csDMARDs such as leflunomide in efficacy [123,136]. The csDMARD, Leflunomide, is metabolized into its pharmacologically active metabolite, A77 1726, which prevents the metabolic needs for clonal expansion and differentiation of effector T cells [137]. Administering low-dose glucocorticoids to csDMARD monotherapy or combination therapies has shown to increase efficacy [100]. However, it is unclear whether immediate combination therapy with different DMARDs is more effective than “step-up” therapy (initial MTX monotherapy with the addition of other csDMARDs if conditions do not improve), although the combination of MTX and etanercept has shown greater efficacy over oral triple therapy with MTX, sulfasalazine, and hydroxychloroquine [123,138]. Hydroxychloroquine diffuses to the lysosome, which is known as the center of immunometabolism due to selective amino acids being stored in the lysosome and it being a crucial signaling hub for mTORC1 responses [139]. Hydroxychloroquine’s presence within the lysosome can lead to an alteration of innate immune cell function [140]. Whereas sulfasalazine affects adaptive immune responses by preventing folate metabolism, resulting in a decrease in TH1 differentiation and TH1 driven cytokine production [18,141]. Although biologics are not yet well understood, etanercept is known to function by binding to TNFα and TNFβ (lymphotoxin) to inhibit pro-inflammatory TNF-mediated responses [18,142]. Interestingly, recent studies suggests that etanercept reduces bone metabolism in patients with an arthritic disease known as ankylosing spondylitis [143]. However, if the target disease activity in RA is not achieved through csDMARDs, bDMARDs, such as TNF-α inhibitors, then targeted synthetic DMARDs (tsDMARDs), such as janus kinase (JAK)-inhibitors, are generally administered in combination with MTX [122]. JAK inhibitors, such as tofacitinib, reduce inflammation by decreasing proinflammatory cytokine signaling and production [144]. Additionally, tofacitinib has shown to be implicated in lipid metabolism but has not shown any effects on glucose metabolism [144,145]. The combination of bDMARDs with MTX was found to improve clinical response and outcomes more than either MTX or bDMARDs alone [146]. However, bDMARDs have several harmful side effects, including an increased risk of non-melanoma skin cancers, gastrointestinal perforation, and pulmonary infections [147]. Similarly, tsDMARDs, namely tofacitinib, have demonstrated high efficacy both as a monotherapy and in combination with MTX [123,148], but side effects include serious infections, reactivation of latent tuberculosis, increases in serum creatinine, and decreased lymphocyte and neutrophil counts [148].
Besides DMARDs, other treatments for RA include nonsteroidal anti-inflammatory drugs (NSAIDs) and corticosteroids. NSAIDs, which include aspirin and ibuprofen, are anti-inflammatory and analgesic agents that block production of prostaglandins (PGs) by inhibiting the enzyme prostaglandin G/H (PGG/H) synthase, also known as COX. This inhibition of PGs is linked to the suppression of immune cell expansion [149]. While NSAIDs are effective in reducing pain and stiffness, they usually do not reduce acute-phase reactants or change the radiographic progression of disease [150]. However, the use of NSAIDs has been associated with altered blood pressure and an increased risk of hypertension [151], and NSAIDs except for naproxen have been shown to increase the risk of myocardial infarction [152]. Additionally, a recent study discovered that NSAIDs, namely ibuprofen, can alter 34 metabolic pathways that are implicated in metabolism of amino acids, hormones, and vitamins as well as production of ROS and hydrogen peroxide intracellularly in male mice [153]. Furthermore, ibuprofen may have a greater impact on the liver than previously believed, however, the impact of ibuprofen in male mice differed from female mice, warranting that further research in understanding drug metabolism as a whole in males versus females should be preformed [153]. Further, corticosteroids which affect all immune cells given their cell surface expression of the glucocorticoid receptor [154], have been proposed as a treatment for RA, but their use has been overshadowed by long-term steroid toxicity and several adverse side effects, including serious infections, gastrointestinal bleeding, and accelerated atherosclerotic vascular diseases [155]. Additionally, treatment with low-dose prednisolone (PRED) alone has been associated with significant bone mineral density (BMD) loss, while the BMD in patients receiving a combined treatment of PRED and MTX was higher but still significantly lower than non-RA controls [156]. A temporary duration of a moderate dose of the corticosteroid prednisone has shown to affect glucose metabolism in healthy subjects with no effects on muscle protein metabolism or function [157]. However, Short et al. argues that a temporary duration of prednisone in healthy patients induces muscle insulin resistance in glucose and amino acid metabolism, with a weakened protein anabolism [158]. Furthermore, a study investigating corticosteroid use in RA patients treated with infliximab found an association between corticosteroid use and increased risk of infection, and consequently, the authors suggested that corticosteroids should be administered for the shortest period possible to achieve remission [159]. Notably, GCs and NSAIDs may interact with metabolic homeostasis, which can lead to CV risk. Nonetheless, GCs, MTX, sulfasalazine and leflunomide effect T-cell functions and modulate pro-inflammatory TH1-driven cytokines while altering TH1/TH2 immune-mediated responses [160]. Although each of these FDA approved RA therapeutics affect immune cells and metabolism (Table 1), it is not fully understood how some may affect immunometabolism, therefore indicting that further research is required. Nonetheless, there is a clear unmet clinical need for RA therapeutics, however, future developments may benefit from designing therapeutics to target the immunometabolism of specific immune cells.
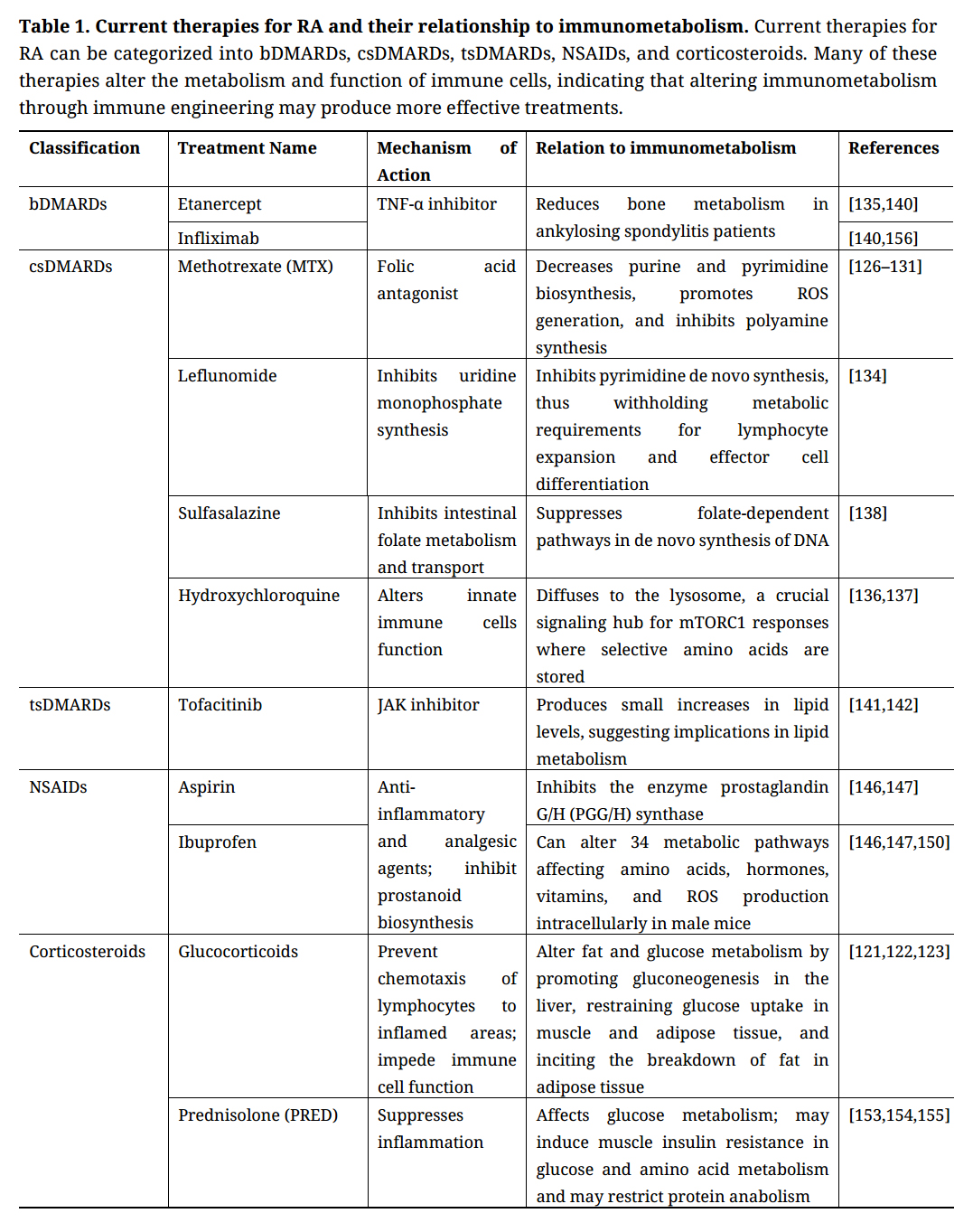 Table 1. Current therapies for RA and their relationship to immunometabolism. Current therapies for RA can be categorized into bDMARDs, csDMARDs, tsDMARDs, NSAIDs, and corticosteroids. Many of these therapies alter the metabolism and function of immune cells, indicating that altering immunometabolism through immune engineering may produce more effective treatments.
Table 1. Current therapies for RA and their relationship to immunometabolism. Current therapies for RA can be categorized into bDMARDs, csDMARDs, tsDMARDs, NSAIDs, and corticosteroids. Many of these therapies alter the metabolism and function of immune cells, indicating that altering immunometabolism through immune engineering may produce more effective treatments.
Among the established RA mouse models, the antigen-induced CIA mouse model remains the most commonly studied due to the model having several pathological similarities to RA in humans as well as the ease associated with CIA induction [161]. Interestingly, RA in humans and CIA in mice can both ultimately lead to chronic and destructive polyarthritis with signs of synovitis and bone and cartilage erosion. This progressive destruction of the articular joints are characterized by fibrin deposition, proliferative synovial cells, periosteal bone development, mononuclear infiltrates and pannus development [162,163]. Moreover, the susceptibility to developing RA in CIA mice is largely facilitated by I-Aq, an major histocompatibility complex (MHC) class II receptor that binds the equivalent immunodominant collagen peptide region as the human RA-associated allele human leukocyte antigen (HLA)-DR4 (DRB1*0401) [164]. Although collagen’s role in initiating RA is not well understood, its localization within the main sites of inflammation in RA demonstrates that its fundamental processes involved in initiating CIA in mice may share similar features with the effector phase of RA in humans [165]. In human RA, collagen is likely not the antigen that initiates the disease, rather it becomes the target antigen in RA as a result of epitope spreading as studies have shown that the lung may be the initial site of autoimmune response in RA patients [166]. Nevertheless, both RA in humans and CIA in mice are exacerbated by the induction of autoreactive B and T cell responses, where B cells exacerbate RA by producing autoantibodies and cytokines and by activating autoreactive T cell responses via co-stimulatory molecules CD80 or CD86 while presenting autoantigens [161,167,168]. Notably, the autoantibody production in the CIA model, illustrated by elevated levels of IgG1 and IgG2a, has made the CIA model desirable as a preclinical RA model for diagnostics and for the development of antibody based therapeutics [167,169]. Therefore, downregulating the metabolism of inflammatory immune cells can reduce symptoms in both human RA and murine CIA.
The ability to understand the specific amino acid sequences of collagen that is recognized by the MHC receptors on DCs in CIA mice has created therapeutic opportunities of two major approaches toward antigen-specific immunotherapy. The first being the disruption of I-Aq-restricted antigen presentation by delivering synthetic collagen peptide analogues containing a dominate T cell epitope region [170]. The second being the delivery of collagen to tolerogenic DCs (tolDCs) for the induction of tolerogenic antigen-specific immune responses [171–174]. Although antigen-specific immunotherapies hold promise for the development of effective RA therapeutics, the autoantigen within RA mouse models and within RA patients may differ. Additionally, autoantigens that are exclusive to RA have yet to be discovered, therefore further research is required to identify autoantigens and the respective patient-specific TCRs in RA. Nonetheless, to maximize the potential of antigen-specific immunotherapies, these therapies should consider the versality of an antigen when generating antigen-specific formulations. Thus, further investigation within antigen-induced arthritis mouse models may contribute to the development of optimal and personalized antigen-specific therapies for RA. Importantly, since a number of autoimmune diseases share similar pathological processes during disease development, the synthesis of a versatile antigen-specific formulation may lead to the generation of a platform that can be applicable to treating other autoimmune diseases [175].
Although current RA therapeutics, such as DMARDs and NSAIDs, suppress inflammatory responses for the treatment of RA symptoms, these therapeutics do not discriminate between RA specific and non-specific immune cells, and therefore, compromise the patient’s immune system. Thus, there is a clear unmet clinical need for RA therapeutics that address specific pro-inflammatory immune responses at the root cause of RA. Nonetheless, extensive preclinical and clinical research has shown the efficacy of immunotherapies and their ability to overcome the challenge in autoimmunity by targeting immune cells that are attacking healthy cells and tissues without causing further damage to the healthy cells and tissues themselves. Therefore, utilizing immunoengineering to target metabolism of pro-inflammatory immune cells may restore immune tolerance in autoimmune diseases such as RA without causing damage to healthy cells and tissues.
Cellular therapies targeting T cells or DCs has shown effective and promising results in preclinical RA studies. As previously discussed, human RA effector T cells are metabolically impaired, however, these effector T cells are still capable of rapidly expanding and secreting inflammatory cytokines, despite their inability to produce quick ATP via the glycolytic pathway [15,28]. Thus, T cells therapies have been explored as an approach to remedy the metabolic dysregulation of human RA T cells. For example, Wright et al. demonstrated that the adoptive transfer of antigen-specific TREGs, induced by the retroviral gene transfer of Foxp3 and/or TCR, lead to the suppression of RA in mice [176], potentially by increasing the ratio of TREGs to metabolically impaired effector T cells. Similarly, the adoptive transfer of ex vivo expanded TREGs and antigen-specific TREGs, extracted from CIA mice with RA, ameliorated RA in CIA mice [177,178]. However, methods to expand TREGs for patients have been restricted by low cell numbers, challenging manufacturing methods, and limited understanding of patient-specific TCRs for the recognition of disease-relevant MHC-peptide complexes [179–181]. Additionally, the stability of ex vivo expanded TREGs remains questionable [182], however, there may be some promise in utilizing clustered regularly interspaced short palindromic repeats (CRISPR) to overcome this hurdle. The BACH2 gene has been found to potentially increase Foxp3 and TREG stability. Additionally, Stub1 is known to be involved in Foxp3 ubiquitination [182]. Therefore, CRISPR may play a critical role in enhancing TREG stability by targeting BACH2 or Stub1. However, studies have not yet explored this field for RA specifically, indicating that further studies are required to confirm the role of CRISPR in increasing the stability of ex vivo expanded TREGs cells for RA therapeutics. Additionally, vitamin A, B9 and C have also been explored as a viable option to increase TREG stability when expanded in an ex vivo setting [183]. Nonetheless, activated B cells with high glycolytic activity are also shown to be implicated in RA pathogenesis, therefore, therapies targeting B cells provides a method to prevent the production of autoantibodies and the progression of RA.
Chimeric antigen receptor T cell (CAR-T cell) therapies, which consists of engineering T cells in an ex vivo setting to express an antigenic receptor of interest, have made a remarkable impact in personalized therapies and cancer treatment in clinic, and also demonstrates potential success in preclinical autoimmune studies. An in vitro RA study demonstrated a reduction in autoreactive B cell subsets by engineering CAR-T cells to express an anti-fluorescein isothiocyanate (FITC) receptor with FITC-labelled citrullinated autoantigen peptide epitopes that were recognized by ligands on autoreactive B cells [184]. Given that B and T cells secrete cytokines, and that cytokines impact metabolic and immune pathways, a modulation of B and T cell populations can lead an altered metabolic environment [185,186]. Another group engineered CAR-expressing TREGs to target a filament protein, citrullinated vimentin (CV), and transduced this construct into human TREGs. Although the functional activity of this construct still needs to be tested in vivo in mice, the CAR-expressing TREGs did show a reaction to CV in RA patients synovial fluid and CV-expressing cells [187]. Therefore, this construct may have success in in vivo mice studies. However, there is little research in CAR-T cell therapies for the treatment of RA, indicating that further studies are required.
Significant progress has also been made in DC immunotherapy, specifically the induction of tolDCs or DCs in an anti-inflammatory state, and their ability to reinstate immune tolerance in RA. Activated, or inflammatory, DCs have increased glucose metabolism [6], thus shifting or inhibiting inflammatory pathways or glycolytic energy requirements within DCs can alter inflammatory phenotypes. A few methodologies for successfully inducing DCs with anti-inflammatory properties include inhibiting factors that promote inflammatory responses, such as NF-κB or costimulatory molecule CD80/CD86 [188–191]. DCs with pro-inflammatory properties have increased CD80/CD86 expression [192] and NF-κB is known to govern metabolic adaptations and is key in linking metabolism to inflammation [191,193]. Also, anti-inflammatory DCs can be induced by engineering DCs to genetically, and continuously, express apoptosis-inducing factors (AIF), such as Indoleamine 2,3-dioxygenase (IDO) or Fas ligand (FasL) [191,194,195], which AIFs are believed to play an anatomical role in cellular redox metabolism [196]. Nonetheless, several pharmacological agents are available to induce DCs with anti-inflammatory properties for the subsequent induction of anti-inflammatory T cell responses. For example, the modulation of DC metabolism can lead to anti-inflammatory effects induced by increasing FasL, transforming growth factor beta (TGF-β), IL-10, vitamin D3, and administering pharmacological agents’ rapamycin (inhibits mTOR), BAY 11-7082 (inhibits NF-кB), and metformin (promotes AMPK, which in turn promotes peroxisome proliferator-activated receptor gamma coactivator 1 (PGC1)). Intracellularly, this can lead to an increase in OXPHOS, FAO, IDO and a decrease in glycolysis. The downregulation of co-stimulatory molecules on these DCs and the subsequent release of IL-10 and ROS result in the inhibition of effector T cells and the proliferation of TREGs for the induction of tolerance (Figure 4) [191]. There is limited research on development of tolDCs in RA clinical trials, however there is one clinical study in RA patients that demonstrated that the intra-articular delivery of autologous tolDCs is safe and capable of reducing inflammatory symptoms, though no immunomodulatory effects were found [197]. However, the main goal of this phase 1 trial was to test the safety of autologous tolDCs. Therefore, further clinical studies would need to be done to test the efficacy of autologous tolDCs for the treatment of RA. Nonetheless, therapies targeting DCs, specifically antigen-specific therapies, are exciting and can lead to a personalized modulation of immune responses in RA.
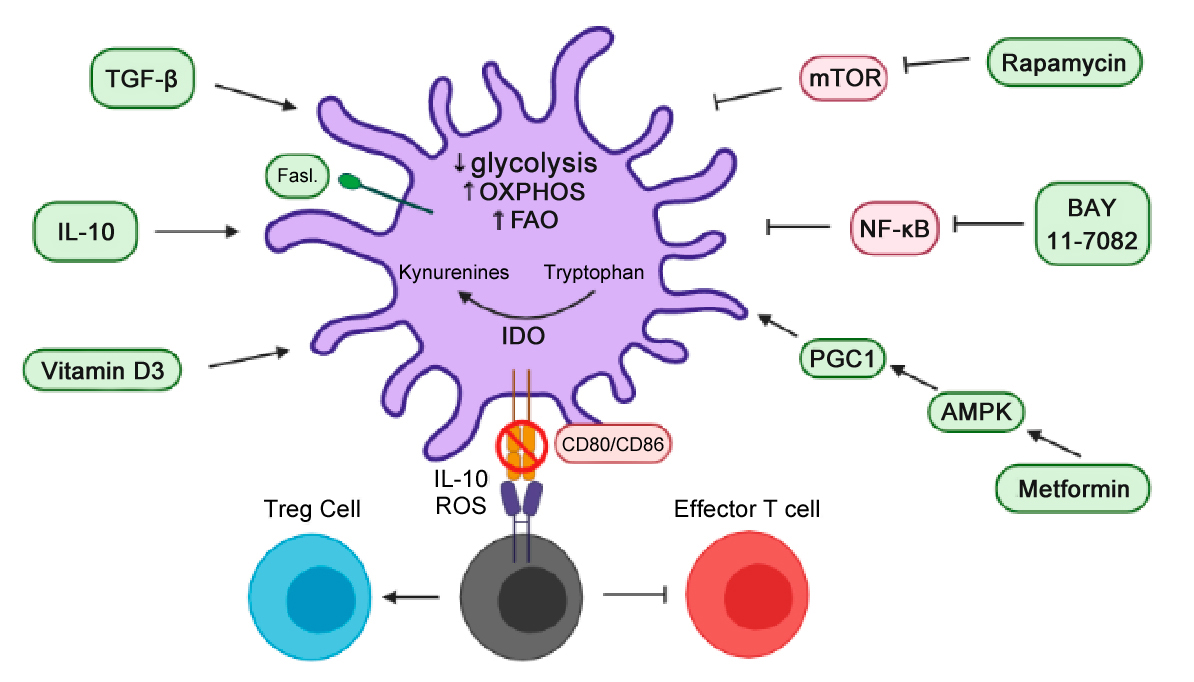 Figure 4. Stimulating anti-inflammatory DCs using cytokines and pharmacological agents. The administration of cytokines, vitamin D3 or pharmacological agents can stimulate an intracellular increase in IDO, OXPHOS, and FAO for a downregulation of co-stimulatory molecules and a subsequent release of IL-10 and ROS. As a result, this can lead to an inhibition of effector T cells and the proliferation of TREGs for the induction of tolerance.
Figure 4. Stimulating anti-inflammatory DCs using cytokines and pharmacological agents. The administration of cytokines, vitamin D3 or pharmacological agents can stimulate an intracellular increase in IDO, OXPHOS, and FAO for a downregulation of co-stimulatory molecules and a subsequent release of IL-10 and ROS. As a result, this can lead to an inhibition of effector T cells and the proliferation of TREGs for the induction of tolerance.
Semi-mature DCs (induced by repeatedly pulsing DCs with TNF-α) that were pulsed with the CIA self-antigen, collagen type II, resulted in antigen-specific responses with an impeded onset of RA and low arthritis scores [173]. TNF-α is traditionally know to induce pro-inflammatory DC responses, however, continuous administration of TNF-α (or a maturation stimuli) can lead to a reduced DC production of pro-inflammatory cytokines, and an increased production of IL-10, therefore inducing a tolDC phenotype with the ability to lead to cytokine-mediated changes within the metabolic environment [172,174]. This induction of tolDCs led a reduction in antigen-specific antibody production and a decreased ability to trigger T cell proliferation [173]. Similarly Popov et al., demonstrated that tolDCs, which were induced by inhibiting NF-κB, were able to modulate antigen-specific B and T cell responses when tolDCs pulsed with a RA self-antigen were injected into CIA mice [171]. However, the translation of antigen-specific therapies to clinic is limited by the ability to identify autoantigens and the respective patient-specific TCRs for effective and personalized results. Therefore, there is an urgent need to generate methodologies to accurately identify autoantigens in RA patients.
Interestingly, the discovery of peptide tetramers has become a useful tool in identifying, characterizing, and eliminating self-reactive B and T cells and their respective autoantigen. In 2003, Altman et al. reported methodologies in generating synthetic TCR ligands containing a peptide sequence with a fluorescent tag for identifying antigen-specific T cells [198]. Similar methodologies utilized this concept for the development of autoantigen-carrying tetramers which were capable of detecting autoreactive B cells [199]. This technology of peptide tetramers has achieved an expansion of peptide-specific CD4+ and CD8+ regulatory T cells for induction of anti-inflammatory antigen-specific responses in CIA mice for the treatment of RA [200,201]. The identification, characterization, and elimination of self-reactive B and T cells and their respective autoantigen, in the presence of immunometabolism techniques, may provide a novel therapeutic approach to treating RA. However, peptide tetramer studies have predominately occurred in preclinical models. Therefore, further studies would need to be performed in clinical trials in order to understand if peptide tetramers can be a form of personalized treatment for RA.
Nonetheless, gene delivery strategies using both viral vectors or non-viral methodologies (e.g., nanoparticles (NPs)) have been useful in engineering the immune system as a personalized approach to treating RA [202]. For example, a single intra-articular delivery of an adenosine-associated virus (AAV) vector encoding the human interferon-β (hIFN-β) gene was shown to produce hIFN-β in RA FLSs for the regulation of inflammation. Importantly, this transgene expression was regulated by a NF-κB promoter, which is only activated during inflammatory flare ups, therefore this allowed for a controlled expression of the transgene [202,203]. As previously discussed, RA FLSs acquire an aggressive phenotype, in turn transitioning RA FLFs into a hyper-metabolic state with increased glycolysis, amino acid metabolism, and protein biosynthesis [13,89]. Therefore, metabolically reprogramming RA FLSs, by modulating NF-κB mediated responses, can lead to a reduction of inflammatory characteristics within the synovium. Gene delivery has also shown to be effective in increasing the levels of the anti-inflammatory cytokine, IL-10, while avoiding the global immunosuppression that is associated with the systemic administration of recombinant IL-10 [204,205]. IL-10 has previously shown to inhibit the expression of glycolytic genes in bone marrow-derived macrophages (BMDMs) and also has shown to inhibit the glycolysis pathway by decreasing the cellular ability to translocate GLUT1 from intracellular vesicles to the cells surface [206]. Interestingly, a single intranasal delivery of an IL-10 plasmid in a RA CIA mouse model was shown to lead to a reduction in bone destruction and joint inflammation. This modulation in disease was suggested to be influenced by the increased local levels of IL-10 causing a reduction of costimulatory molecules on APCs, in turn reducing T cell activation, T cell mediated IFN-γ production, and macrophage activation [205,207–209]. Although expressing a gene of interest can induce immunosuppressive results, it is vital to ensure genes are delivered in a targeted manner to avoid the immunoactiviting properties associated with immune cells identifying extracellular DNA as damage‐associated molecular patterns (DAMPs) or pathogen‐associated molecular patterns (PAMPs) [210]. Recent studies have shown that continuous exposure to extracellular DNA or increased concentrations of extracellular DNA may contribute to autoimmunity and immune dysregulation [210–212].
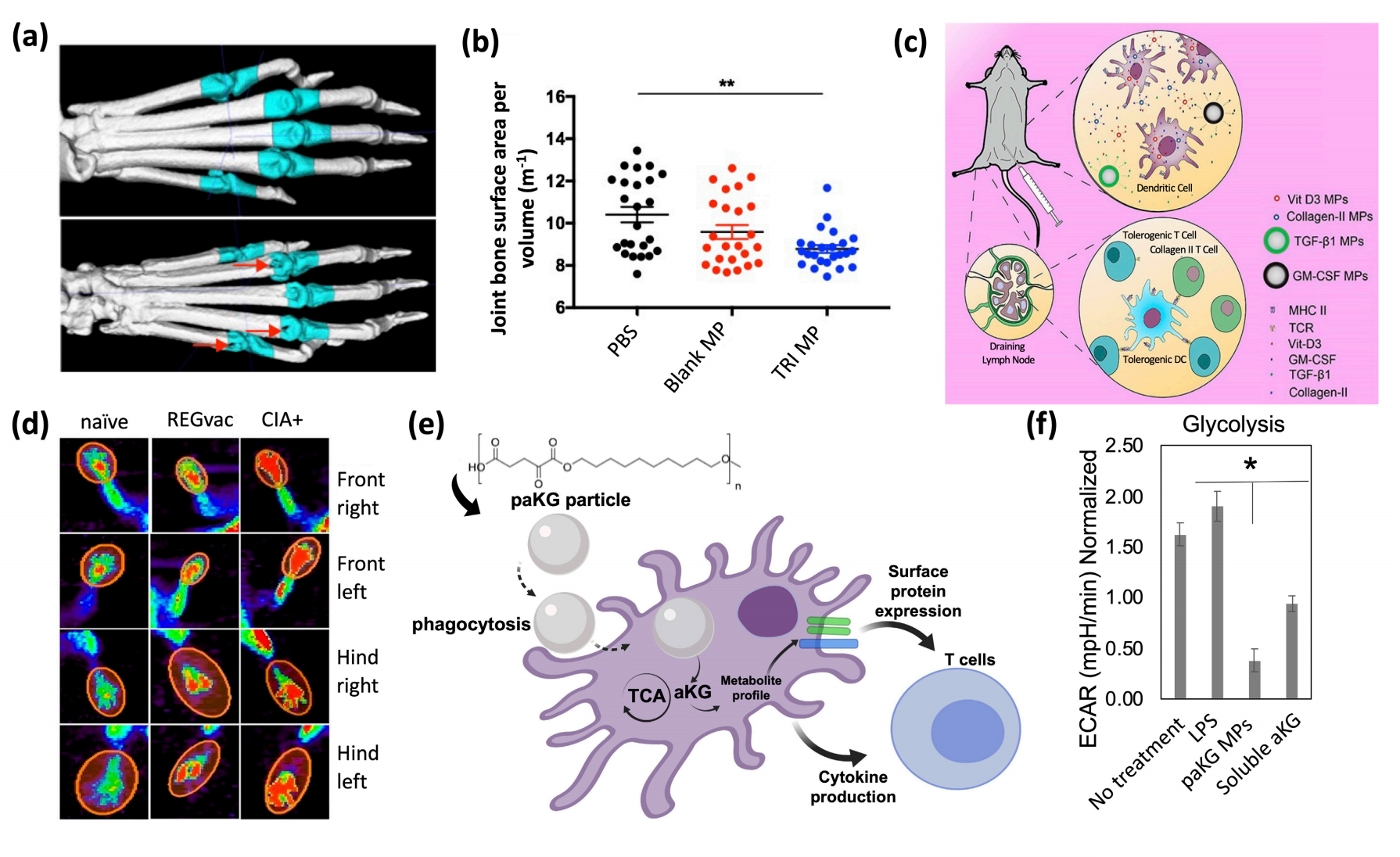 Figure 5. Biomaterials for the modulation of immune responses. (a,b) Relation between TRI MPs and bone erosion, (a) representative micro-CT images for a comparison of a murine paw without bone erosion (top) and an arthritic murine paw with severe bone erosion (bottom), (b) average joint bone surface area to volume ratio to compare, PBS vs blank MPs vs TRI MPs. Reprinted with permission from [213] an open access article distributed under Creative Commons Attribution License. (c,d) Schematic and PET images of regulatory vaccine, (c) schematic of dual-sized REGvac MPs for the treatment of CIA mice, (d) Representative PET images of mice paws 56 days after initial MP treatment. Reprinted with permission from [214]. copyright © 2019 American Chemical Society. (e,f) Schematic and extracellular flux assays of metabolite-based polymeric MPs, (e) schematic of metabolite-based MPs modulating DC responses and subsequent T cell responses, (f) metabolite-based MP-mediated modulation of glycolysis (ECAR = extracellular acidification rate) within DCs. Reprinted with permission from [215]. Copyright © 2020 Royal Society of Chemistry.
Figure 5. Biomaterials for the modulation of immune responses. (a,b) Relation between TRI MPs and bone erosion, (a) representative micro-CT images for a comparison of a murine paw without bone erosion (top) and an arthritic murine paw with severe bone erosion (bottom), (b) average joint bone surface area to volume ratio to compare, PBS vs blank MPs vs TRI MPs. Reprinted with permission from [213] an open access article distributed under Creative Commons Attribution License. (c,d) Schematic and PET images of regulatory vaccine, (c) schematic of dual-sized REGvac MPs for the treatment of CIA mice, (d) Representative PET images of mice paws 56 days after initial MP treatment. Reprinted with permission from [214]. copyright © 2019 American Chemical Society. (e,f) Schematic and extracellular flux assays of metabolite-based polymeric MPs, (e) schematic of metabolite-based MPs modulating DC responses and subsequent T cell responses, (f) metabolite-based MP-mediated modulation of glycolysis (ECAR = extracellular acidification rate) within DCs. Reprinted with permission from [215]. Copyright © 2020 Royal Society of Chemistry.
Although there is a need to enhance the safety and efficacy of immunotherapies, recent efforts have shown that tissue-specific and/or cell-specific immunomodulation can be achieved in a more safe and effective manner when immunoengineering concepts are applied [216,217]. Preclinical RA studies have predominately utilized biomaterials for targeted specificity and for controlling the release rate of immunomodulators to increase their therapeutic window. Poly(D,L-lactic-co-glycolic-acid) (PLGA) is a widely used polymer for encapsulation and drug delivery [218]. Recent efforts have shown that PLGA microparticles (MPs) encapsulating immunomodulatory agents, can lead to the alteration of DC and T cell phenotypes in RA mice. Bassin et al. encapsulated TGF-β, rapamycin, and IL-2 within PLGA MPs (termed as TRI MPs) and recognized a modulation in CD4+ T cell populations with an increase in TREG infiltration within inflamed paws for a reduction of RA severity in CIA mice. On average, mice treated with TRI MPs demonstrated the least amount of bone erosion, as observed by micro-CT imaging (Figure 5a,b) [213]. The proposed mechanism of this technology entailed of decreasing T cell proliferation (due to the release of TGF-β and rapamycin), and the expansion of a regulatory cell population (due to the release of rapamycin, TGF-β and IL-2) [213]. Although TGF-β’s impact on metabolism is not well understood, rapamycin is a widely used mTOR inhibitor and is a master regulator of cell growth and metabolism [219], and IL-2 is a key regulator of T cell metabolism [220]. Furthermore, PLGA MPs can also be utilized in antigen-specific therapies. Allen et al. encapsulated a DC chemoattractant (GM-CSF), vitamin D3, TGF-β1, and the respective RA-relevant autoantigen within PLGA MPs (termed “regulatory vaccine” (REGvac)) for an alteration in inflammatory DC phenotypes, and subsequent inflammatory cytokine production and subsequent inflammatory T cell responses for an antigen-specific treatment of RA in CIA mice (Figure 5c) [214]. The delivery of GM-CSF, vitamin D3 and TGF-β1 can alter the PI3K pathway to allow for the generation of a tolerogenic environment that encourages the differentiation and maintenance of a tolerance-inducing DC phenotype [214,221]. The representative PET images exhibited a significantly lower standard uptake value of glucose in CIA mice treated with REGvac (Figure 5d) [214]. Additionally, Mangal et al. demonstrated that polymeric MPs can be synthesized from an anti-inflammatory Krebs cycle metabolite, alpha-ketoglutarate (aKG, termed poly alpha-ketoglutarate (paKG) MPs), to modulate DC function by altering their energy metabolism for a reduction in glycolytic activity with applications in immune-mediated diseases (Figure 5e,f) [215]. Notably, other biomaterials such as black phosphorus nanosheets (BPNs) within a platelet-rich plasma (PRP)-chitosan thermosensitive hydrogel has shown to treat RA in mice by increasing the levels of ROS within cells of inflamed joints for the elimination of abnormal synovial cells and tissue [222]. A reduction in joint swelling, cartilage damage and inflammatory cytokine production has also been achieved in CIA rats with an intra-articular injection of a thermosensitive hydrogel containing polyethyleneimine NPs that encapsulated and released Indomethacin (IND) and MTX. The delivery of IND with MTX has previously been shown to increase the absorption of MTX [223,224]. Importantly, the use of biomaterials for drug delivery can increase the favorability of immunotherapies by increasing the ease of drug administration and reducing the side effects of off-site targeting. However, biomaterials for the treatment of RA have not yet been implemented in clinic, and these technologies need to be further explored in clinical trials prior to its use as a treatment in RA patients.
Evidence suggests that the dysfunctional humoral and cellular responses in RA are also shared with a number of other autoimmune diseases such as SLE, type 1 diabetes mellitus (T1DM) and MS [175]. Therefore, it is worth exploring immunotherapies that have shown benefit in other autoimmune diseases, and apply them as a potential therapeutic for RA. For example, CD19-CAR T cell therapy, for the elimination of B cells in SLE, has shown to eradicate autoantibody production, resolve disease in target organs, and increase murine lifespan [225]. Considering that in RA B-cell mediated auto-antibody production leads to progression of RA, the depletion of B cells using CD19-CAR T cell therapy may reverse autoantibody production and reduce RA symptoms. Furthermore, given that B and T cells produce cytokines, which are known to impact metabolic and immune pathways, an alteration of B and T cell frequencies can lead modulated metabolic environment [185,186]. Interestingly, in T1DM, CRISPR was utilized to counteract immune rejection when transplanting genetically engineered human embryonic stem cells (hESCs) differentiated insulin-producing cells that were deficient in the β2-microglobulin (B2M) gene, an essential constituent of MHC-I, but expressing a program death-ligand 1 (PD-L1) transgene to prevent the attack by autoreactive T-cells [226]. This concept of genetically editing differentiated hESCs holds translational potential for the treatment of RA as an immune evasive tactic against autoreactive immune cells. On another note, in MS, the first case report for adoptive transfer of autologous Epstein–Barr virus (EBV)-specific T cells had demonstrated an improvement in MS symptoms as observed by MRI for a decrease in disease activity as well as a reduction in IgG production [227]. Since EBV infects 95% of the global population and RA patients have flawed EBV-specific CD8+ T cells, the removal of accumulated EBV-infected B cells, by the adoptive transfer of autologous EBV-specific CD8+ T cells, may lead to the alleviation of RA symptoms in patients who have previously been infected with EBV [228–230]. Furthermore, a preclinical myelin-specific immunotherapy against four myelin basic proteins (MBP) for MS had shown to induce TREGs in the spleen as well as persistent tolerance to the MBPs [231]. The administration of this formulation, which consisted of these four MBP peptides, to MS patients led to a reduction in MS lesions with effects lasting post-treatment [232]. Although a human clinical trial of citrullinated peptide DC immunotherapy for RA has been attempted [233], anti-CCP are autoantigens that are not restricted to RA [234], and therefore, antigen-specific immunotherapies for RA in clinic are limited to an uncertainty in relevant-autoantigens that are specific to RA. However, identification of RA relevant-autoantigens may lead to the similar results as observed in the clinical MS study.
In addition to the therapies that have proven effective in other autoimmune diseases, metabolically engineering immune cells of RA patients offers the potential for permanent and targeted immunoregulation. Six major metabolic pathways associated with immune cell function are glycolysis, the Krebs cycle, the PPP, FAO, FAS and amino acid metabolism [235]. Therefore, rate-limiting enzymes or metabolites within these metabolic pathways represent an ideal target to induce long-term tolerance or potentially remission. As previously noted, T cells transition toward anabolic metabolism once activated and shift from utilizing nutrients for survival and homeostasis toward clonal expansion and effector functions in its activated state [236]. However, memory T cells rely on catabolic metabolism to generate ATP by breaking down glucose, fatty acids, and amino acids to fuel the Krebs cycle and OXPHOS [237]. Thus, the differences in bioenergetic pathways for activated T cells and memory T cell depicts a window for target-based therapies that can reduce off-target effect. For example, mTOR plays a principal role in regulating memory CD8+ T cell differentiation and inhibition of mTOR has been found to prevent glycolysis [238]. The administration of rapamycin, an mTOR inhibitor, in mice with an acute lymphocytic choriomeningitis virus infection had shown to promote memory CD8+ T cell expansion and function. Pan et al. demonstrated that the inhibition of mitochondrial free fatty acid β-oxidation reduces the frequency of memory CD8+ T cells therefore supporting the evidence of memory T cells relying on catabolic metabolism [239]. Though increasing the frequency of memory T cells enables an approach to induce long term tolerance, it is also important to explore increasing the persistence and lifespan of memory T cells for the treatment of RA.
Interestingly, the increased expression of CD39 in memory T cells has been identified as a characteristic of immunosenescence and therefore is implicated in facilitating apoptotic pathways for a decreased ability in mounting effective memory responses [240]. Memory T cells with a high CD39 expression are shown to be metabolically stressed with increased AMPK phosphorylation, decreased cytoplasmic ATP levels, and reduced glucose influx. Taking these characteristics into consideration, in addition to the compromised mitochondrial function of CD39+ memory T cells, this suggests that these cells undergo apoptosis due to the possibility of inadequately producing ATP via OXPHOS. Individuals with low CD39 expression demonstrated an increase in vaccine-specific memory T cells for a more efficacious vaccine response. Pharmaceuticals targeting pAMPK can prevent CD39+ T cells from initiating the apoptotic pathway for an increased durability in memory T cell responses [241]. Therefore, the inhibitory effects of mTOR and AMPK can metabolically reprogram memory T cells for an increased frequency and lifespan to potentially induce long-term tolerance or remission in RA. Nonetheless, pharmaceuticals with metabolic and immunomodulatory effects would need to be delivered in a controlled manner to prevent non-specific immune modulation as described below.
Biomaterials are an effective approach to delivering immunomodulatory agents to cells of interest. The ability to deliver mTOR or AMPK inhibitors in a targeted manner to anti-inflammatory RA-specific T cells may induce extended periods of immune tolerance in RA. Additionally, the delivery of other pharmaceuticals, such as glycolytic inhibitors, to specific immune cells may induce desired effects in targeted cells and prevent non-specific immunomodulation. For example, the delivery of glycolytic inhibitors, such as 2-DG and PFK15, to activated and inflammatory immune cells can lead to a shift toward anti-inflammatory bioenergetic requirements [242]. However, the systemic administration of these small molecules can lead to non-specific glycolytic inhibition and can affect the host’s ability to mount an immune response against other pathogens [243–245]. Therefore, utilizing micro- or nano- particles such as PLGA, for the delivery of these glycolytic inhibitors can lead to a targeted and controlled release [246,247]. Both the size and composition of particles directly affects the release rate of the encapsulated content as well as the overall degradation rate of the particle, thus generating a customizable system [248,249]. Generally, particles with a diameter less than 10 µm can be phagocytosed by APCs for a modulation of their cellular characteristics [250]. PLGA is a highly favored polymer due to its easily adjustable hydrophobicity, biocompatibility and inert characteristics [249]. However, delivering immunomodulatory agents within an anti-inflammatory particle for the treatment of RA may synergize the targeted immune cells’ ability to shift toward an anti-inflammatory state. As mentioned previously, Mangal et al. demonstrated that an anti-inflammatory metabolite-based MP can be synthesized from the Krebs cycle metabolite aKG for an induction of non-activated DC phenotype and an upregulation in metabolic pathways associated with immune suppression [215]. Therefore, utilizing this technology as a delivery vehicle for inflammatory diseases, such as RA, may be impactful in generating a targeted anti-inflammatory response as compared to utilizing an inert particle as a delivery vehicle. Remarkably, the delivery of paKG MPs encapsulating a glycolytic inhibitor, PFK15, and the collagen-induced arthritis self-antigen, collagen type II, led to suppression in DC activation and a subsequent generation of immunosuppressive antigen-specific responses for an ultimate normalization in paw inflammation in CIA mice [251]. Biomaterials may also be utilized to modify the function of a number of immune cells implicated in RA, and therefore, should be studied in cell types such as APCs, T cells, neutrophils, ILCs and FLSs. Furthermore, Colombo et al. demonstrated that particles coated with a synovium homing peptide can lead to a targeted delivery of particles within synovial tissue [252]. Therefore, synovium homing peptides are an additional methodology that can be employed to prevent non-specific immunomodulation. Although there are several metabolic aberrations within adaptive immune cells, innate immune cells, and within the synovial environment, several immunoengineering approaches have been discovered for the modulation of metabolic abnormalities for the treatment of RA symptoms (Figure 6). Nevertheless, further preclinical and clinical research is required to continue advancing and generating safe, effective and personalized therapeutics for RA.
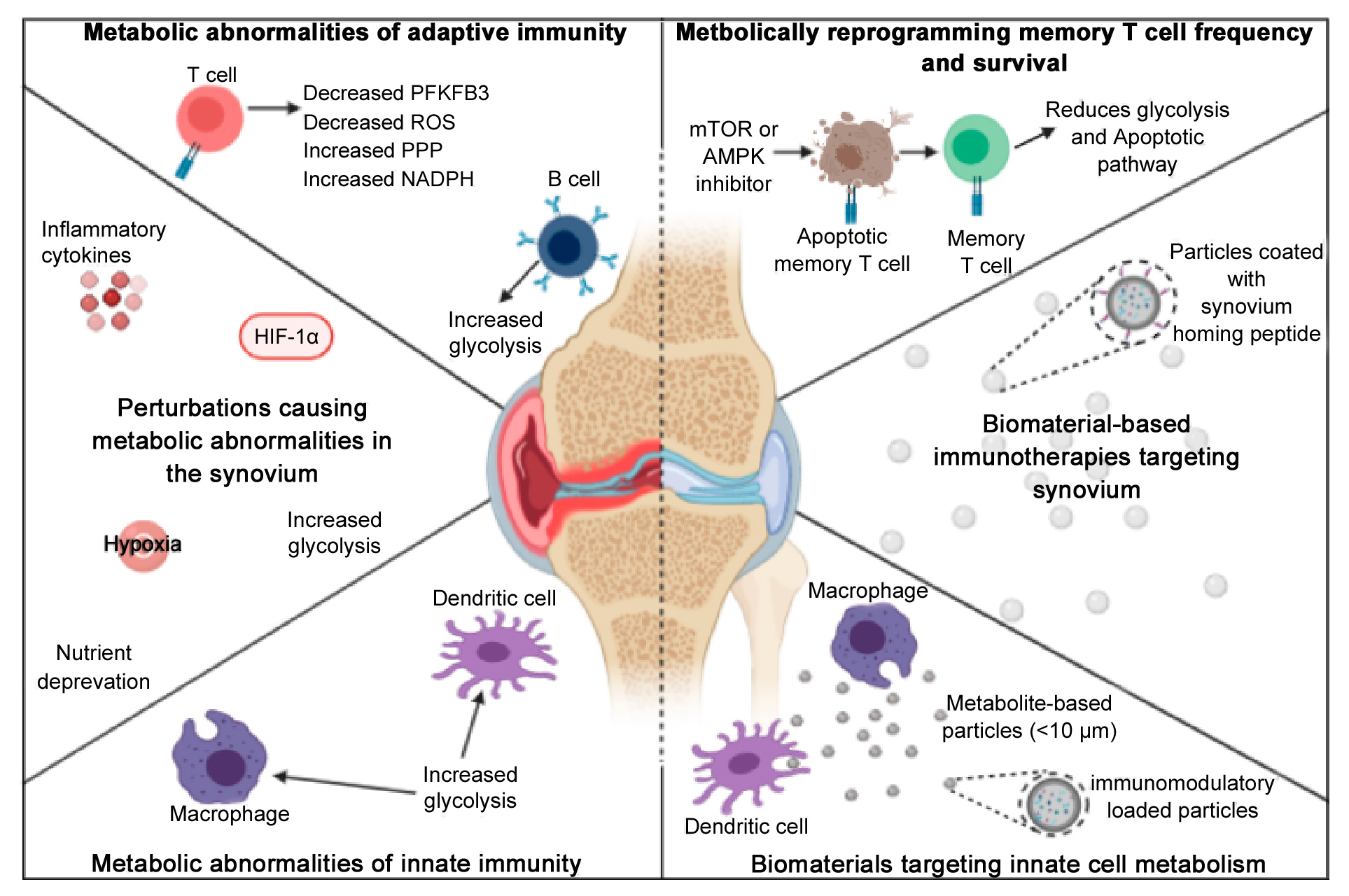 Figure 6. Metabolic abnormalities within RA and immunoengineering approaches for the modulation of metabolism. Metabolic abnormalities (right side) within adaptive immune cells, the synovium and innate immune cells have shown to lead to the progression of RA and synovial inflammation. Several immunoengineering approaches (right side) have been discovered for the modulation of metabolic abnormalities for the treatment of RA symptoms.
Figure 6. Metabolic abnormalities within RA and immunoengineering approaches for the modulation of metabolism. Metabolic abnormalities (right side) within adaptive immune cells, the synovium and innate immune cells have shown to lead to the progression of RA and synovial inflammation. Several immunoengineering approaches (right side) have been discovered for the modulation of metabolic abnormalities for the treatment of RA symptoms.
RA is a complex and metabolically heterogeneous disease, in turn making it difficult to treat with standard therapeutics. The finding of metabolic irregularities contributing to immune dysregulation in autoimmunity has led to the identification of novel therapeutic strategies. As such, the pairing of immunometabolism approaches with concepts of immunoengineering may lead to the development of more effective therapeutics by being able to target specific metabolic pathways. As shown in this review, biomaterials, such as micro- and nano- particles, can be synthesized from an anti-inflammatory metabolite to generate an immunosuppressive delivery vehicle capable of targeting and modulating phagocytes such as DCs. Importantly, micro- and nano- particles can encapsulate various immunomodulatory agents to induce desired effects in targeted immune cells to induce long-term tolerance or remission. However, the joint field of immunometabolism and immunoengineering face a challenge of formulating precise delivery systems that are capable of enhancing therapeutic function of encapsulated molecules that are negatively charged or hydrophilic, while circumventing nonspecific toxicities, off-target effects and undesired pharmacokinetics and low bioavailability [216]. Furthermore, the field is challenged by scale-up manufacturing for the translation of biomaterial-based immunotherapy to clinic for the treatment of RA [216]. Therefore, further research is required prior to the clinical translation of biomaterials with immunometabolism reprogramming capabilities for the treatment of RA.
The authors declare that they have no conflicts of interest.
This research was funded by NIH grant R01 AR078343, “Local immunometabolism modulating biomaterials for immunosuppressive applications”.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
80.
81.
82.
83.
84.
85.
86.
87.
88.
89.
90.
91.
92.
93.
94.
95.
96.
97.
98.
99.
100.
101.
102.
103.
104.
105.
106.
107.
108.
109.
110.
111.
112.
113.
114.
115.
116.
117.
118.
119.
120.
121.
122.
123.
124.
125.
126.
127.
128.
129.
130.
131.
132.
133.
134.
135.
136.
137.
138.
139.
140.
141.
142.
143.
144.
145.
146.
147.
148.
149.
150.
151.
152.
153.
154.
155.
156.
157.
158.
159.
160.
161.
162.
163.
164.
165.
166.
167.
168.
169.
170.
171.
172.
173.
174.
175.
176.
177.
178.
179.
180.
181.
182.
183.
184.
185.
186.
187.
188.
189.
190.
191.
192.
193.
194.
195.
196.
197.
198.
199.
200.
201.
202.
203.
204.
205.
206.
207.
208.
209.
210.
211.
212.
213.
214.
215.
216.
217.
218.
219.
220.
221.
222.
223.
224.
225.
226.
227.
228.
229.
230.
231.
232.
233.
234.
235.
236.
237.
238.
239.
240.
241.
242.
243.
244.
245.
246.
247.
248.
249.
250.
251.
252.
Mangal JL, Basu N, Wu H-JJ, Acharya AP. Immunometabolism: An Emerging Target for Immunotherapies to Treat Rheumatoid Arthritis. Immunometabolism. 2021;3(4):e210032. https://doi.org/10.20900/immunometab20210032

Copyright © 2021 Hapres Co., Ltd. Privacy Policy | Terms and Conditions




