Location: Home >> Detail
J Psychiatry Brain Sci. 2025;10(6):e250016. https://doi.org/10.20900/jpbs.20250016

Department of Obstetrics, Gynecology and Reproductive Sciences, University of Maryland School of Medicine, Baltimore, MD 21201, USA
Schizophrenia (SCZ) is a complex neuropsychiatric disorder characterized by disruptions in cognition, perception, and social behavior. While traditional research has focused on neurotransmitter dysregulation, growing evidence implicates altered lipid metabolism as a key contributor to the disease’s molecular pathology. Lipids are essential for maintaining neuronal membrane integrity, facilitating synaptic transmission, supporting myelination, and regulating neuroinflammation, all processes disrupted in SCZ.
This review examines the molecular alterations in lipid pathways in SCZ, focusing on the dysregulated metabolism of phospholipids, sphingolipids, cholesterol, and polyunsaturated fatty acids (PUFAs). We highlight findings from genetic studies, neuroimaging, and patient-derived induced pluripotent stem cell (iPSC) models, which collectively provide a translational framework for studying lipid-related abnormalities. Evidence from postmortem brain tissue and peripheral samples consistently reveals altered lipid profiles in individuals with SCZ.
Integrative models combining genetic risk variants with environmental stressors such as maternal immune activation and perinatal hypoxia offer deeper insights into how lipid dysregulation emerges during neurodevelopment and impairs neuronal function. From a clinical perspective, targeting lipid metabolism presents promising avenues for biomarker discovery, early diagnosis, and therapeutic innovation. Lipid-based interventions, including targeting of sphingolipid or phosphatidylinositol pathways, may be particularly beneficial for treatment-resistant cases.
By integrating molecular lipidomics with patient-specific models and systems-level approaches, this review underscores the potential of lipid-focused strategies to advance our understanding of SCZ pathophysiology and guide the development of novel treatments.
SCZ is a severe mental disorder characterized by dysregulated cognition, emotion, and behavior that affects approximately 1% of the global population. It results from the interaction of genetic and environmental factors [1–4]. Much classical research has focused on neurotransmitters, particularly dopamine, glutamate, and risk factors and associated genes [5–8]. Genome-wide association (GWAS) studies have identified copy number variants, single-nucleotide variations, related pathways, risk factors, and genes that confer susceptibility. More evidence now points to significant metabolic dysfunction as a central component and possibly a causative feature of the disorder [9,10]. Individuals with SCZ often exhibit high rates of metabolic abnormalities, including insulin resistance, altered lipid profiles, mitochondrial dysfunction, and systemic inflammation, even before antipsychotic treatment [11–14]. These findings have led to a paradigm shift, recognizing that SCZ is not only a brain-based disorder but also a metabolic syndrome with neuropsychiatric features. Disruptions in glucose and lipid metabolism may contribute to the disease’s underlying mechanisms and impact brain development, synaptic plasticity, and neurotransmitter systems involving dopamine and glutamate. Understanding the bidirectional link between brain function and peripheral metabolic health is essential, as it offers new opportunities for early detection, mechanistic understanding, and treatment strategies. This metabolic perspective may ultimately unify various clinical features of SCZ and reshape its management as a neuro-metabolic disorder.
Lipid metabolism is an important but often overlooked aspect of how SCZ develops. Lipids are essential for brain growth, membrane health, synaptic function, and myelination, all of which are impacted in individuals with SCZ [15–17]. Changes in lipid profiles, including imbalances in phospholipids, sphingolipids, and cholesterol, are consistently observed in blood samples and postmortem brain tissue of affected people [18]. These changes may contribute to neuroinflammation, oxidative stress, and disrupted signaling in neurotransmitter systems like dopamine and glutamate. Furthermore, antipsychotic medications can worsen lipid imbalances, adding complexity to the disorder’s metabolic profile. Recent advances in lipidomics and systems biology are beginning to reveal how lipid metabolic pathways interact with genetic and environmental risk factors, offering new insights into disease mechanisms [15,19]. Therefore, lipid metabolism is not just a consequence of treatment or lifestyle but may be a fundamental biological pathway underlying both the cognitive and physical features of SCZ. Understanding this lipid–brain connection could lead to new biomarkers for early diagnosis and novel targets for treatment.
The purpose of this review is two-fold: first, to highlight and provide a perspective on the progress in lipid metabolic alterations in the etiology and progression of SCZ, and secondly, to underline that potential disease-modifying and treatment strategies may involve a metabolic component. This review draws upon the seminal works of many laboratories worldwide, and its goal is to provide new directions for future work in the altered lipid landscape in SCZ.
Metabolism and Psychiatric DisordersMetabolic homeostasis is crucial for brain health, and increasing evidence indicates that its dysfunction is closely linked to the development of psychiatric disorders. The emerging field of metabolic psychiatry bridges the connection between metabolic balance and mental well-being, blending the principles of biological psychiatry and optimal metabolic function [20–23]. Changes in glucose and lipid metabolism, mitochondrial dysfunction, and systemic inflammation have been observed in conditions such as SCZ, bipolar disorder, and depression, often independent of medication effects [11,12,14]. These metabolic issues can interfere with neuronal signaling, synaptic plasticity, and neurodevelopmental processes, contributing to cognitive and emotional symptoms (Figure 1).
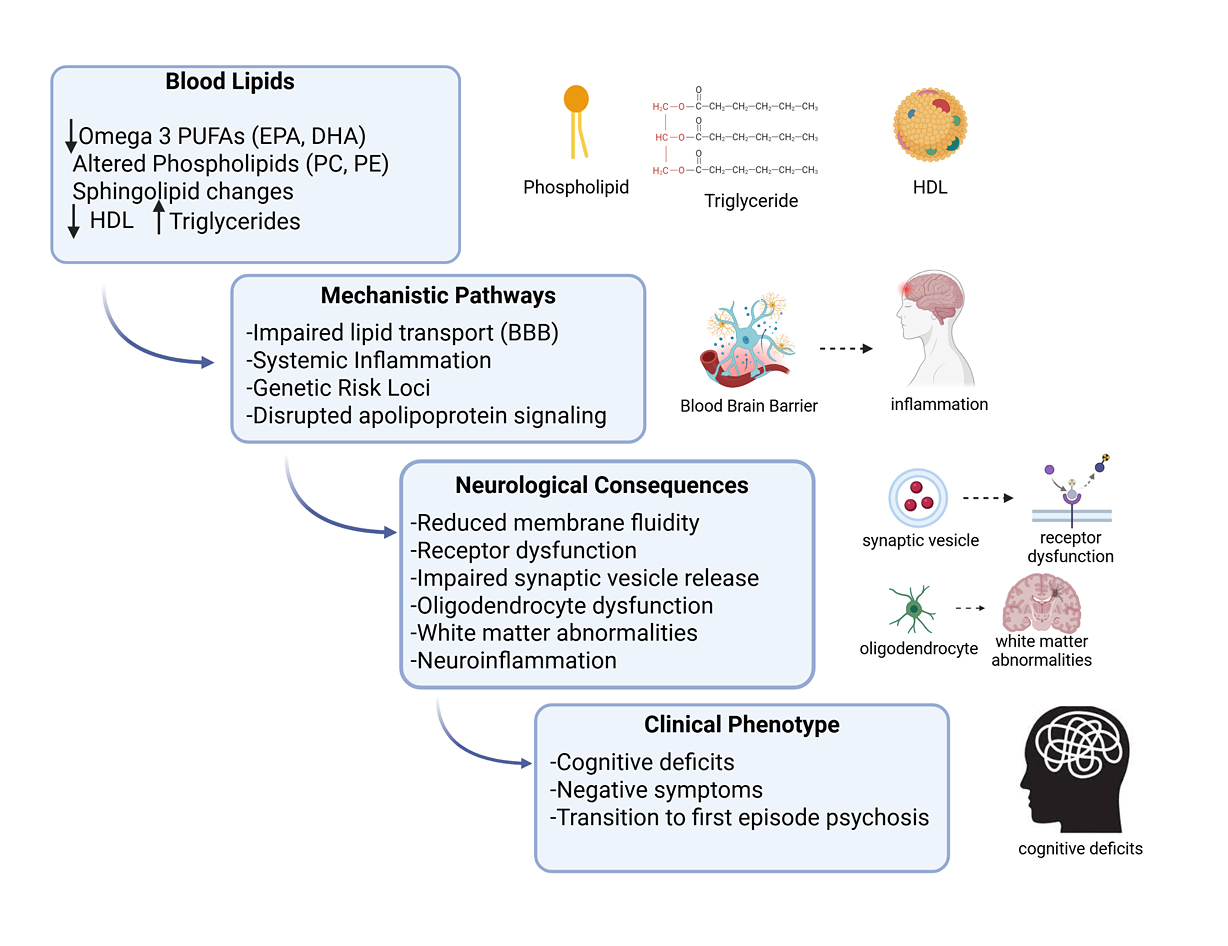 Figure 1. Peripheral lipid dysregulation as a precursor to psychosis. A schematic illustration shows how peripheral lipid abnormalities may contribute to the onset of psychosis. Blood lipids: Reduced omega-3 polyunsaturated fatty acids (PUFAs) (EPA, DHA), altered phospholipids (phosphatidylcholines, phosphatidylethanolamines), sphingolipid changes, and dyslipidemia (↓ HDL cholesterol, ↑ triglycerides). Mechanistic pathways include impaired lipid transport across the blood–brain barrier, systemic inflammation, genetic risk loci regulating lipid metabolism, and disrupted apolipoprotein signaling. Neurological consequences involve reduced membrane fluidity and receptor dysfunction, impaired synaptic vesicle release, oligodendrocyte dysfunction and white matter abnormalities, and increased neuroinflammatory signaling. The clinical phenotype includes these molecular and cellular disturbances manifesting as cognitive deficits, negative symptoms, and the transition from high-risk states to first-episode psychosis. HDL = High-Density Lipoprotein, EPA = Eicosapentaenoic Acid, DHA = Docosahexaenoic Acid. Arrows pointing downward (↓) indicate downregulation, while arrows pointing upward (↑) indicate upregulation. [Created in BioRender. Roychaudhuri, R. https://BioRender.com/xz3oi1m accessed on 10 Nov 2025].
Figure 1. Peripheral lipid dysregulation as a precursor to psychosis. A schematic illustration shows how peripheral lipid abnormalities may contribute to the onset of psychosis. Blood lipids: Reduced omega-3 polyunsaturated fatty acids (PUFAs) (EPA, DHA), altered phospholipids (phosphatidylcholines, phosphatidylethanolamines), sphingolipid changes, and dyslipidemia (↓ HDL cholesterol, ↑ triglycerides). Mechanistic pathways include impaired lipid transport across the blood–brain barrier, systemic inflammation, genetic risk loci regulating lipid metabolism, and disrupted apolipoprotein signaling. Neurological consequences involve reduced membrane fluidity and receptor dysfunction, impaired synaptic vesicle release, oligodendrocyte dysfunction and white matter abnormalities, and increased neuroinflammatory signaling. The clinical phenotype includes these molecular and cellular disturbances manifesting as cognitive deficits, negative symptoms, and the transition from high-risk states to first-episode psychosis. HDL = High-Density Lipoprotein, EPA = Eicosapentaenoic Acid, DHA = Docosahexaenoic Acid. Arrows pointing downward (↓) indicate downregulation, while arrows pointing upward (↑) indicate upregulation. [Created in BioRender. Roychaudhuri, R. https://BioRender.com/xz3oi1m accessed on 10 Nov 2025].
The human brain is rich in lipids, with about 50% of its dry weight made up of various types, broadly categorized as phospholipids, sphingolipids, cholesterol, and fatty acids. Phospholipids form the lipid bilayer of membranes, featuring a polar headgroup and a nonpolar tail. Sphingolipids are a distinct class characterized by a sphingosine backbone and are involved in maintaining membrane integrity and signaling. Cholesterol contains a sterol nucleus, a hydrocarbon tail, and a hydroxyl group, serving as a key component of cell membranes and helping maintain fluidity. These molecules are essential for the structural stability of neuronal membranes and processes like synaptic transmission, myelination, and intracellular signaling. Brain lipids are crucial for myelination, the process by which oligodendrocytes or Schwann cells form myelin sheaths around axons. Cholesterol provides rigidity and structural stability to the myelin membrane. It is essential for membrane compaction and proper myelin layering. Phospholipids (e.g., phosphatidylcholine, phosphatidylethanolamine) form the basic bilayer structure of myelin membranes and contribute to membrane fluidity and signaling. Horrobin proposed that SCZ is a disorder of membrane lipid metabolism, and an abnormality of membrane lipids affects neurological functions and complex brain behaviors [10,24]. Sphingolipids (e.g., sphingomyelin, galactosylceramide, sulfatide) are crucial for membrane stability and signaling. Galactosylceramide and sulfatide, major glycosphingolipids unique to myelin, play roles in membrane adhesion and compaction. These major classes of lipids play critical roles in signal transduction, the process by which cells convert external signals into internal responses. Lipids are therefore not just passive membrane components; they actively initiate, coordinate, and propagate cellular signals [25–28] (Figure 2).
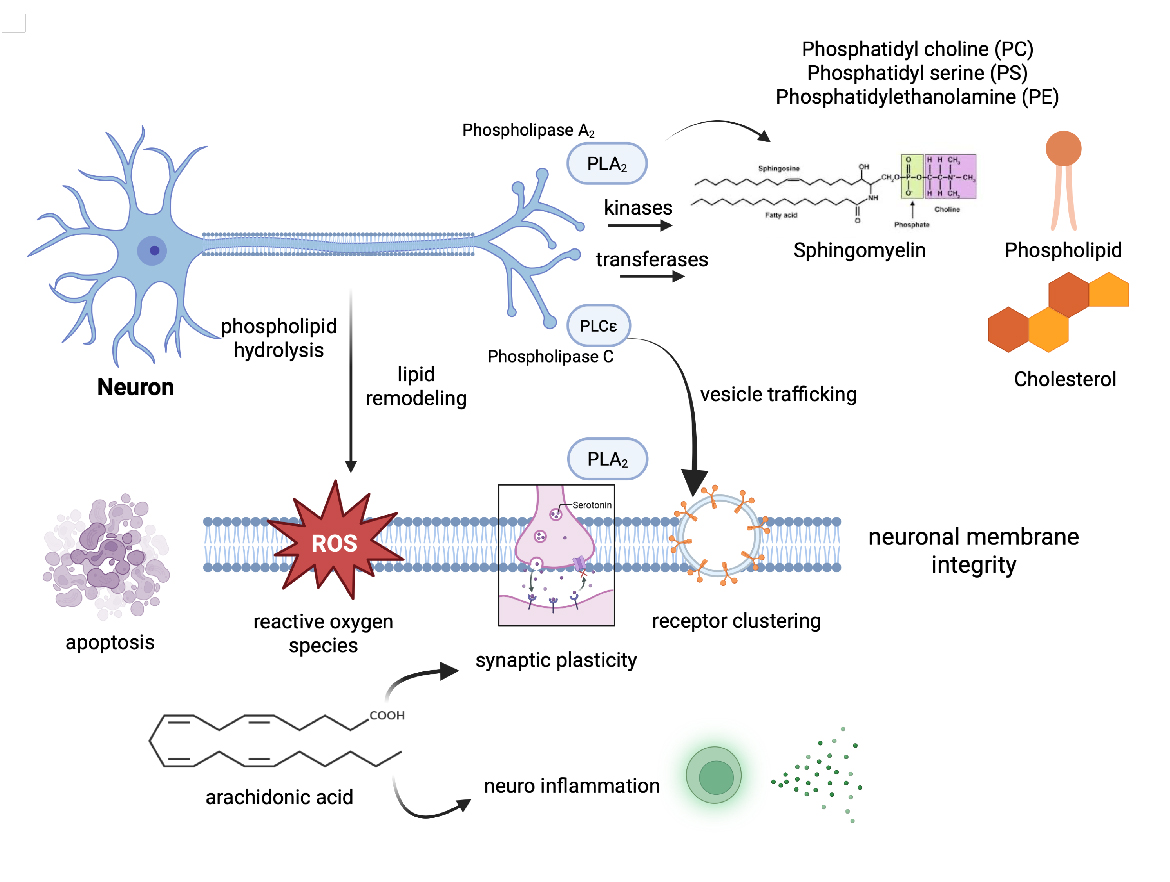 Figure 2. Phospholipid Metabolism and Membrane Integrity in Neurons. This schematic illustrates key phospholipid metabolic pathways and their roles in maintaining neuronal membrane integrity. Phosphatidylcholine (PC), phosphatidylethanolamine (PE), and phosphatidylserine (PS) are produced and remodeled via enzymatic reactions involving phospholipases (PLA, PLC, PLD), kinases, and transferases. Phospholipase-driven hydrolysis releases bioactive lipids like arachidonic acid, which can activate signaling pathways influencing neuroinflammation and synaptic function. Preserving PS asymmetry and membrane fluidity is essential for vesicle trafficking, receptor clustering, and synaptic plasticity. Disruption of these pathways may weaken membrane integrity, increase vulnerability to oxidative stress and apoptosis, and contribute to neuropsychiatric disorders. [Created in BioRender. Roychaudhuri, R. https://BioRender.com/a462kr1 accessed on 10 Nov 2025].
Figure 2. Phospholipid Metabolism and Membrane Integrity in Neurons. This schematic illustrates key phospholipid metabolic pathways and their roles in maintaining neuronal membrane integrity. Phosphatidylcholine (PC), phosphatidylethanolamine (PE), and phosphatidylserine (PS) are produced and remodeled via enzymatic reactions involving phospholipases (PLA, PLC, PLD), kinases, and transferases. Phospholipase-driven hydrolysis releases bioactive lipids like arachidonic acid, which can activate signaling pathways influencing neuroinflammation and synaptic function. Preserving PS asymmetry and membrane fluidity is essential for vesicle trafficking, receptor clustering, and synaptic plasticity. Disruption of these pathways may weaken membrane integrity, increase vulnerability to oxidative stress and apoptosis, and contribute to neuropsychiatric disorders. [Created in BioRender. Roychaudhuri, R. https://BioRender.com/a462kr1 accessed on 10 Nov 2025].
Phospholipids are key components of cellular membranes and act as precursors for signaling molecules. Phosphatidylinositol (PI) and its derivatives (e.g., PIP2, PIP3) play a central role in intracellular signaling pathways. PIP2 is cleaved by phospholipase C (PLC) into Inositol trisphosphate (IP3) → releases Ca2⁺ from the ER. Diacylglycerol (DAG) → activates Protein Kinase C (PKC). PIP3, produced by PI3K, activates the Akt/PKB pathway (cell growth, survival). Phosphatidylserine (PS) anchors signaling proteins (like PKC) to membranes via electrostatic interactions. It plays a role in apoptotic signaling when exposed on the outer membrane leaflet [29–31].
Sphingolipids, especially sphingomyelin and glycosphingolipids, are structural and signaling molecules. Ceramide is a central hub in sphingolipid metabolism and is involved in stress response, apoptosis, and cell cycle arrest [32–34]. Sphingosine-1-phosphate (S1P) regulates cell migration, vascular maturation, and immune cell trafficking and binds to S1P receptors (G-protein-coupled receptors (GPCRs)) on the cell surface [35,36]. Glycosphingolipids (e.g., gangliosides) modulate receptor activity and participate in neurodevelopment and immune responses [37,38]. Cholesterol organizes the plasma membrane and forms lipid rafts, microdomains that concentrate signaling molecules. Lipid rafts facilitate receptor clustering (e.g., T-cell receptors, growth factor receptors) and enhance the efficiency and specificity of signal transduction. It influences the function of GPCRs and ion channels [39,40] (Figure 2).
Due to the restrictive properties of the blood–brain barrier, the brain’s relative independence from peripheral lipid pools underscores the importance of intrinsic lipid regulatory mechanisms within neurons and glia. Perturbations in these homeostatic processes may not only underlie neurodevelopmental vulnerabilities but also contribute to disease progression and treatment response in SCZ.
The concept of lipid homeostasis, the precise balance between lipid synthesis, degradation, and intercellular transport, is critical for sustaining normal brain function.
Human studies consistently emphasize lipid dysregulation as a reliable and clinically important feature of SCZ. Advances in lipidomics have revealed widespread changes in circulating and tissue lipid profiles, implicating disruptions in phospholipid, sphingolipid, and cholesterol metabolism [15]. These abnormalities are observed across various biological matrices, including plasma, serum, cerebrospinal fluid (CSF), and postmortem brain tissue, indicating that lipid imbalance is not confined to one compartment but reflects systemic and central disturbances [41]. These changes seem to be independent of antipsychotic treatment, suggesting they may be inherent disease mechanisms rather than side effects of medication. Collectively, these findings highlight the importance of lipidomics research in understanding the metabolic origins of SCZ and in identifying new biomarkers with diagnostic and therapeutic potential.
Plasma and serum studies provide the most direct evidence for systemic lipid abnormalities in SCZ [15,42]. Numerous reports have documented reduced levels of PUFAs, altered phosphatidylcholine and phosphatidylethanolamine species, and shifts in cholesterol and triglyceride profiles. These alterations often correlate with negative symptoms and cognitive impairment, suggesting functional relevance beyond metabolic health (Figure 1). Importantly, several lipidomic changes have been observed in antipsychotic-naïve patients, reinforcing the view that dyslipidemia may be a core component of the disorder rather than solely a treatment consequence [43,44]. Together, plasma and serum data highlight lipid dysregulation as both a potential biomarker reservoir and a window into the systemic manifestations of SCZ.
CSF analyses provide a more direct reflection of central nervous system biochemistry, and studies have identified significant lipid alterations in SCZ. Reductions in phosphatidylserine, sphingomyelin, and specific PUFAs have been reported, consistent with impaired membrane integrity and altered neurotransmission [24,42,45–47]. These findings point to disturbances in lipid metabolism at the brain level, complementing systemic signatures observed in blood. Although sample sizes in CSF studies remain modest due to limited availability, the reproducibility of lipid abnormalities underscores their potential as central biomarkers of disease processes.
Postmortem investigations have provided critical insights into region-specific lipid dysregulation in SCZ. Alterations in phospholipids, sphingolipids, and cholesterol metabolites have been identified in cortical and subcortical regions, implicating disrupted lipid metabolism in synaptic dysfunction and myelin integrity [17,42,46,47]. These abnormalities often overlap with pathways identified in plasma and CSF, reinforcing the systemic central link in lipid pathology. Importantly, postmortem lipidomics also highlights heterogeneity across brain regions, suggesting that localized metabolic imbalances may contribute to the diverse symptom dimensions of SCZ. Collectively, these studies strengthen the hypothesis that lipid dysregulation is not merely epiphenomenal but central to disease pathophysiology.
Taken together, evidence from plasma, serum, CSF, and postmortem brain tissue converges on the view that lipid dysregulation is a robust and multidimensional feature of SCZ. The consistency of abnormalities across peripheral and central compartments suggests that lipid imbalance is not an isolated metabolic disturbance but a systemic hallmark of the disorder. These findings strengthen the rationale for integrating lipidomic biomarkers into clinical research, both to improve early detection and to guide personalized interventions. Moreover, the overlap between peripheral and central lipid signatures raises the possibility that accessible biofluids, such as plasma, could serve as practical surrogates for brain pathology, bridging mechanistic insight with translational utility. The following are a few classes of lipids altered in the onset of psychosis.
Polyunsaturated Fatty Acids (PUFAs)Several studies have shown reduced levels of omega-3 and omega-6 PUFAs in individuals at risk for psychosis. Lower levels of Eicosapentaenoic Acid (EPA) and Docosahexaenoic Acid (DHA), which are essential components of neuronal membranes, have been linked to a higher chance of developing psychosis in prospective cohorts. These deficiencies may impair membrane fluidity, neurotransmitter receptor function, and anti-inflammatory signaling, potentially leading to early cognitive and behavioral changes. Supporting this, intervention studies indicate that supplementing omega-3 fatty acids in ultra-high risk (UHR) individuals can decrease the transition rates to psychosis. However, results from larger trials have been inconsistent [28,48,49] (Figure 1).
Phospholipids and SphingolipidsLipidomic profiling has revealed abnormalities in phosphatidylcholines, phosphatidylethanolamines, and sphingomyelins in both UHR and first episode psychosis (FEP) populations [15,50,51]. Altered phospholipid species may impair vesicle formation, synaptic transmission, and intracellular signaling cascades. Moreover, disturbances in sphingolipid metabolism have been linked to white matter abnormalities and oligodendrocyte dysfunction, processes increasingly recognized as central to SCZ pathophysiology (Figure 1) [52,53].
Cholesterol and LipoproteinsAbnormalities in cholesterol metabolism have been reported before antipsychotic treatment. Reduced high-density lipoprotein (HDL) cholesterol and elevated triglycerides have been observed in FEP patients, with some studies suggesting an association with negative symptom severity and cognitive deficits [54]. Given cholesterol’s essential role in synapse formation and myelination, even modest deviations may have significant neurodevelopmental consequences [55]. The involvement of apolipoproteins such as ApoE further highlights the intersection of lipid transport pathways and genetic susceptibility to psychosis (Figure 1)[56].
Mechanistic Links Between Peripheral and Central Lipids.The connection between circulating lipid profiles and central nervous system homeostasis remains an area of active investigation [15]. Proposed mechanisms include impaired lipid transport across the blood–brain barrier, peripheral inflammatory states that perturb lipid metabolism, and shared genetic regulation of lipid pathways and SCZ risk loci. While direct causality remains to be established, converging evidence suggests that blood lipid abnormalities may mirror or even drive central metabolic disruptions relevant to psychosis (Figure 1) [50,57].
Clinical Implications of Lipid Dysregulation.The reproducibility of lipidomic signatures in pre-psychotic and FEP populations positions blood lipid profiles as promising biomarkers for early detection and risk stratification. In addition, they raise the possibility of novel therapeutic interventions targeting lipid metabolism, such as dietary supplementation, pharmacological modulation of lipid pathways, or anti-inflammatory strategies. However, heterogeneity across cohorts, small sample sizes, and potential confounding factors such as diet and lifestyle emphasize the need for large-scale, longitudinal, and multi-omic studies [58–62].
The biological effects of lipid imbalance go beyond metabolic comorbidity and provide possible insights into the mechanisms underlying SCZ. Lipids are crucial for maintaining neuronal membrane integrity, synaptic vesicle movement, and receptor activity, and their disruption may directly affect neurotransmission and neural connectivity [63,64]. For instance, disturbances in phospholipids and sphingolipids can impair membrane fluidity and signal transduction, while cholesterol imbalance impacts synapse formation and myelination [17,65]. Additionally, oxidative stress and inflammation, both common in SCZ, can worsen lipid peroxidation, further harming neuronal function [46,65–67]. These mechanisms collectively suggest that lipid disruption is not just an innocent bystander but a potential key player in the neurobiology and symptoms of SCZ (Figure 2).
Lipids are essential for brain development, where they guide neuronal migration, dendritic growth, and synapse formation [37,68]. Disruption of lipid availability or composition during critical developmental windows may interfere with synaptic pruning, a process already implicated in SCZ risk. Abnormal pruning, driven in part by altered lipid-mediated signaling, could lead to excessive synapse elimination and contribute to long-term deficits in neural circuitry and cognition [7,69].
Phospholipids and PUFAs regulate receptor function and intracellular signaling cascades for key neurotransmitters. Altered lipid rafts can impair dopamine receptor trafficking and glutamate receptor clustering, weakening synaptic signaling [70,71]. These disruptions may help explain how lipid imbalance converges on dopaminergic hyperactivity and glutamatergic hypofunction, the two core neurochemical hypotheses of SCZ.
Lipid dysregulation is tightly coupled to inflammatory and redox pathways [72,73]. PUFAs, which comprise neuronal membranes, are substrates for pro-inflammatory eicosanoids, and oxidative stress readily triggers lipid peroxidation, generating toxic byproducts that damage membranes and proteins [28]. Evidence of increased lipid peroxidation in SCZ supports a model in which metabolic imbalance and oxidative injury converge to exacerbate neural dysfunction [74,75]. Reactive carbonyl species (RCS), such as methylglyoxal, 3-deoxyglucosone, and 4-hydroxynonenal (4-HNE), are also produced during oxidative stress and lipid peroxidation. These reactive intermediates generate advanced glycation and lipoxidation end-products (AGEs/ALEs), which interfere with membrane and cytoskeletal functions. Elevated serum AGEs, including pentosidine and carboxymethyl-lysine, have been found in individuals with SCZ before starting antipsychotic treatment, indicating that carbonyl stress might be an inherent pathogenic factor in the disorder [76] (Figure 3).
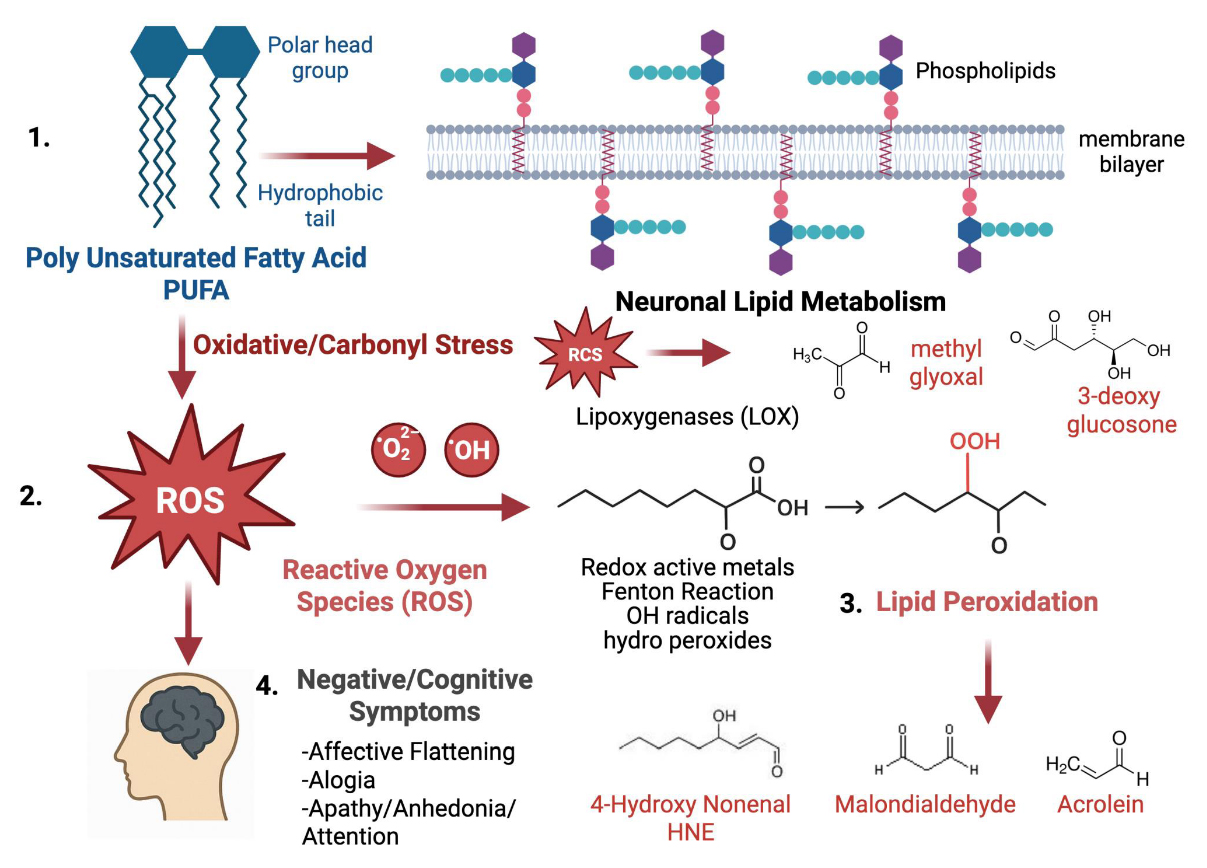 Figure 3. Oxidative stress causes lipid peroxidation and affects neuronal function. Polyunsaturated fatty acids (PUFAs) are vital parts of neuronal phospholipid membranes, helping maintain their structure and supporting signaling functions. During oxidative and carbonyl stress, reactive oxygen species (ROS) and reactive carbonyl species (RCS), such as superoxide, hydroxyl radicals, methyl glyoxal, and 3-deoxyglucosone, attack lipids containing PUFAs, initiating lipid peroxidation through enzymatic (lipoxygenases) and non-enzymatic (redox-active metals, Fenton Reaction) pathways. These reactions produce highly reactive aldehydes, including 4-hydroxy-2-nonenal (HNE), malondialdehyde (MDA), and acrolein, which weaken membrane stability and cause more oxidative damage. The buildup of lipid peroxidation products increasingly links to neuronal dysfunction and the development of negative symptoms in neuropsychiatric disorders [1]. [Created in BioRender. Roychaudhuri, R. https://BioRender.com/wcbzvp6 accessed on 10 Nov 2025].
Figure 3. Oxidative stress causes lipid peroxidation and affects neuronal function. Polyunsaturated fatty acids (PUFAs) are vital parts of neuronal phospholipid membranes, helping maintain their structure and supporting signaling functions. During oxidative and carbonyl stress, reactive oxygen species (ROS) and reactive carbonyl species (RCS), such as superoxide, hydroxyl radicals, methyl glyoxal, and 3-deoxyglucosone, attack lipids containing PUFAs, initiating lipid peroxidation through enzymatic (lipoxygenases) and non-enzymatic (redox-active metals, Fenton Reaction) pathways. These reactions produce highly reactive aldehydes, including 4-hydroxy-2-nonenal (HNE), malondialdehyde (MDA), and acrolein, which weaken membrane stability and cause more oxidative damage. The buildup of lipid peroxidation products increasingly links to neuronal dysfunction and the development of negative symptoms in neuropsychiatric disorders [1]. [Created in BioRender. Roychaudhuri, R. https://BioRender.com/wcbzvp6 accessed on 10 Nov 2025].
Lipids also serve as key energy substrates and regulators of mitochondrial activity. Abnormalities in fatty acid utilization and mitochondrial lipid composition have been implicated in reduced bioenergetic efficiency in SCZ. These disruptions may compromise neuronal resilience and contribute to synaptic deficits [67].
Collectively, these mechanistic insights suggest that lipid dysregulation in SCZ is not confined to a single pathway but reflects a multifaceted influence spanning development, neurotransmission, and cellular resilience. Disruptions in lipid composition can compromise membrane integrity, derail neurodevelopmental processes such as synaptic pruning, and alter dopaminergic and glutamatergic signaling, thereby influencing core symptoms (Figure 3). In parallel, deficits in myelination, heightened oxidative stress, and impaired mitochondrial function point to converging pathways that weaken neural connectivity and plasticity. Rather than being secondary to antipsychotic exposure or metabolic comorbidity, lipid imbalance may represent a primary vulnerability that interacts with genetic and environmental factors to shape the course of illness. Framing lipid disruption within this integrative model highlights its potential both as a biomarker of disease processes and as a novel target for therapeutic intervention.
Dyslipidemia is a highly prevalent metabolic abnormality in individuals with SCZ, manifesting as elevated triglycerides, reduced HDL cholesterol, and variable alterations in low-density lipoprotein (LDL) cholesterol and total cholesterol [77,78]. While antipsychotic medications, particularly second-generation agents, are well-recognized contributors to adverse lipid profiles, a growing body of evidence suggests that dyslipidemia is not solely a treatment-related phenomenon. Altered lipid levels have been observed in antipsychotic-naïve patients and those experiencing a FEP, implicating intrinsic metabolic disturbances in the disorder [54,79]. Dyslipidemia may reflect disruptions in lipid transport, apolipoprotein signaling, or systemic inflammation, all of which converge on pathways critical for brain lipid homeostasis. Importantly, dyslipidemia has been linked to cognitive impairment, negative symptom severity, and increased cardiovascular morbidity, underscoring its dual relevance to both psychiatric and somatic health outcomes [50,54,77]. The interplay between dyslipidemia and SCZ pathophysiology remains incompletely understood, but emerging lipidomic and genetic studies point toward shared mechanisms that may underlie both metabolic and neuropsychiatric phenotypes.
Neurons heavily depend on an external supply of lipids. They have limited ability to synthesize PUFAs and rely on astrocytes for essential fatty acids and cholesterol [80–82]. Neuronal lipids mainly support membrane excitability, receptor function, and synaptic vesicle turnover. Astrocytes function as the metabolic hub for lipid processing. They import dietary precursors, perform elongation and desaturation of fatty acids using enzymes like Fatty Acid Desaturases (FADS 1 and 2) and Elongation of Very Long Chain Fatty acids (ELOVLS), and package PUFAs and cholesterol into Apo E-containing lipoproteins for neuronal delivery [83–86]. Lipidomic studies show decreased DHA and Arachidonic Acid (AA) content in neuronal membranes, affecting NMDA receptor activity, dopamine signaling, and synaptic plasticity [64,87,88]. Increased lipid peroxidation and oxidative stress make neuronal membranes particularly vulnerable [10,89].
While some transcriptomic data from astrocytes show downregulation of FADS1/2 and lipid transport genes, impairing PUFA synthesis and lipid shuttling [90–92], there is no clear evidence in astrocytes from SCZ postmortem tissue that lipid transport genes (e.g., ABCA1, ABCG1: ATP Binding Cassette Sub Family A Member 1, ATP Binding Cassette Sub Family G Member 1) are significantly downregulated in a way clearly tied to impaired PUFA synthesis or shuttling, though there are changes in cholesterol-related genes (SNAP: Synaptic Neuron and Astrocyte Program). Overactive astrocytic AA metabolism promotes pro-inflammatory prostaglandins and leukotrienes, fueling neuroinflammation. Studies have shown that deficient release of DHA-rich lipids weakens neuronal resilience and synaptic repair [93–95].
Functional Consequences of Neuron–Astrocyte Lipid Crosstalk Synaptic DysfunctionNeurons deprived of astrocyte-derived PUFAs show disrupted receptor signaling and impaired neurotransmission [96,97].
NeuroinflammationAstrocytic overproduction of ω-6-derived eicosanoids may amplify microglial activation, while reduced ω-3-derived mediators diminish anti-inflammatory tone [98,99].
Energy MetabolismImpaired astrocyte–neuron lipid trafficking contributes to metabolic inflexibility, a recurring feature in SCZ [10,86].
Therapeutic and Biomarker ImplicationsBoth neuronal and astrocyte metabolism and their crosstalk provide avenues for therapeutic interventions and have implications for biomarker discovery.
-
-
Exosome-based biomarkers: Distinguishing neuron-derived vs astrocyte-derived vesicle lipid signatures could provide cell-type-specific readouts of SCZ pathology.
Advances in genomics and epigenomics have begun to illuminate the heritable and regulatory components linking lipid metabolism to SCZ. GWAS studies have identified risk loci in genes involved in lipid synthesis, transport, and remodeling, suggesting that inherited perturbations in lipid homeostasis may contribute to disease susceptibility [100–104]. Complementary epigenetic analyses reveal that DNA methylation, histone modifications, and noncoding RNAs can regulate the expression of lipid-related genes in the brain and periphery, potentially influencing both developmental trajectories and adult neuronal function [105,106]. Betaine-dependent one-carbon metabolism directly regulates phosphatidylcholine synthesis through the PEMT (phosphatidylethanolamine N-methyltransferase) and BHMT (betaine-homocysteine methyltransferase) pathways. Recent studies have shown that SCZ is associated with reduced plasma betaine and impaired BHMT–PEMT activity, resulting in abnormal lipid methylation, oxidative imbalance, and NMDA receptor hypofunction. In Kif3b⁺/⁻ mice (Kinesin family 3B), reduced KIF3-dependent CRMP2 (Collapsin Response Mediator Protein 2) trafficking leads to neuronal and behavioral deficits, which are rescued by betaine supplementation through restoration of CRMP2 transport [107,108]. Integrating these genetic and epigenetic findings with lipidomic profiles offers a powerful framework to understand how inherited and environmentally responsive mechanisms converge to shape lipid disturbances in SCZ, providing insights into both pathophysiology and potential therapeutic targets.
Genetic Variants in Lipid Metabolism GenesAccumulating evidence suggests that common and rare genetic variants in lipid metabolism genes contribute to SCZ susceptibility. Among the most studied is APOE, which encodes apolipoprotein E, a key regulator of cholesterol transport and lipid homeostasis in the brain. Variants in APOE, particularly the ε4 allele, have been associated not only with altered lipid profiles but also with cognitive deficits and brain structural changes relevant to SCZ [8,109–111]. Similarly, genes involved in fatty acid desaturation, such as FADS1 and FADS2, which regulate the biosynthesis of long-chain PUFAs, show associations with both altered plasma lipid composition and SCZ risk [112–114]. Disruption of these pathways may influence membrane fluidity, neurotransmission, and neuroinflammation.
Transcriptional regulators of lipid metabolism, including SREBF1 (sterol regulatory element-binding transcription factor 1), also harbor variants linked to SCZ. While SREBF1 controls the expression of genes involved in cholesterol and fatty acid synthesis, and its dysregulation can impact neuronal membrane composition, myelination, and synaptic function, there is one report on a myelin-related gene, which is contradictory [100,105,115,116]. Collectively, these genetic findings support a model in which inherited variation in lipid metabolism genes shapes individual vulnerability to SCZ, potentially by modulating both systemic lipid profiles and brain-specific lipid-dependent processes.
Transcriptomic and Methylation Evidence for Lipid Pathway DysregulationEmerging transcriptomic studies in SCZ provide compelling evidence that lipid metabolism is disrupted at the gene expression level [6,46,117]. Analyses of postmortem brain tissue, peripheral blood, and patient-specific iPSC derived neurons reveal altered expression of genes involved in phospholipid synthesis, fatty acid elongation, cholesterol biosynthesis, and sphingolipid metabolism [46,118,119]. For instance, downregulation of SREBF1, FADS1/2, and PEMT has been reported, suggesting that both membrane composition and lipid signaling pathways may be compromised in SCZ [10,113,120]. These transcriptional alterations often correlate with neuroanatomical changes, cognitive deficits, or metabolic comorbidities, highlighting their functional relevance.
Epigenetic modifications further reinforce the role of lipid pathway dysregulation. DNA methylation profiling in brain and blood samples indicates hyper or hypomethylation at promoters and enhancers of lipid-related genes, potentially altering their transcriptional activity [121–124]. Histone modifications and noncoding RNAs, including microRNAs targeting lipid metabolism transcripts, have also been implicated in modulating lipid homeostasis in SCZ [125–128]. Together, transcriptomic and epigenetic evidence suggest that lipid pathway disruption arises from both genetic predisposition and dynamic regulatory processes, providing mechanistic insight into how systemic and central lipid abnormalities may develop in the disorder.
Collectively, genetic variants, transcriptomic changes, and epigenetic modifications all impact lipid metabolic pathways in SCZ, emphasizing a multi-layered framework for disease risk. Inherited variants in genes like APOE, FADS, and SREBF1 may make individuals more likely to have altered lipid levels, while abnormal gene expression in neurons and peripheral tissues reflects both underlying disease mechanisms and adaptive responses. Epigenetic mechanisms, such as DNA methylation, histone modifications, and microRNA regulation, further influence lipid-related gene activity, potentially connecting environmental factors to metabolic disruptions. This integrated view highlights lipid dysregulation as a key aspect of SCZ that results from the interaction of genetic vulnerability and dynamic regulatory processes, offering potential biomarkers and new therapeutic targets.
Understanding the complex interplay between lipid metabolism and SCZ requires robust experimental models that recapitulate key aspects of the disorder. SCZ is a multifactorial condition with genetic, environmental, and neurodevelopmental contributions, and accordingly, preclinical models are diverse, encompassing pharmacological, genetic, and environmental approaches [129–131].
Pharmacological ModelsPharmacologically induced models typically involve the administration of compounds that perturb neurotransmitter systems implicated in SCZ, such as dopamine or glutamate. For instance, the administration of N-methyl-D-aspartate (NMDA) receptor antagonists (e.g., ketamine, phencyclidine) induces behavioral and cognitive deficits reminiscent of the negative and cognitive symptoms of SCZ [132,133].
Non-human primates (NHPs) (especially macaques, marmosets) have prefrontal cortex and other brain structures more similar in cytoarchitecture, connectivity, and developmental timeline to humans than rodents. This provides relevance in modelling higher cognitive functions (working memory, context processing, social cognition). NHPs provide better translational read outs for Electro Encephalogram/Event Related Potential (ERP) signals and correspond more directly with human ERP oscillation biomarkers (e.g., Mismatch Negativity, P3a oscillation, Auditory Steady State Response), enabling comparison of neural signatures. These models also alter lipid composition in the brain, including membrane phospholipids and sphingolipids, highlighting potential mechanistic links between neurotransmission dysregulation and lipid homeostasis [134–137].
Genetic ModelsGenetic manipulations provide insight into SCZ-associated susceptibility genes and their effects on lipid metabolism. Mouse models targeting genes such as DISC1 (Disrupted in Schizophrenia1), COMT (Catechol-O-Methyl Transferase), NRG1 (Neuregulin1), and SRR (Serine Racemase) exhibit behavioral abnormalities relevant to SCZ, as well as alterations in lipid signaling pathways, including sphingolipid and phosphatidylinositol metabolism [138–144]. While mouse models do not recapitulate the core symptoms of SCZ, they are particularly useful for studying the developmental trajectory of lipid dysregulation in synaptic and neuronal deficits [145].
Environmental and Neurodevelopmental ModelsEnvironmental stressors, such as prenatal immune activation or perinatal hypoxia, are widely used to model the developmental origins of SCZ [146–151]. Maternal immune activation models, induced by poly (Inosinic: Cytidylic acid) or lipopolysaccharide exposure during gestation, produce offspring with behavioral phenotypes akin to SCZ, accompanied by perturbations in brain lipid composition and oxidative stress markers [152]. These findings support the notion that early-life disruptions in lipid metabolism may contribute to disease pathophysiology.
Integrated ApproachesIncreasingly, combinatorial models that integrate genetic susceptibility with environmental insults provide a more comprehensive recapitulation of SCZ pathology [153–155]. Such models are valuable for examining how lipid metabolism intersects with neurodevelopmental and neurotransmitter pathways to influence disease onset and progression.
Overall, these experimental models offer a critical platform to investigate the role of lipids in SCZ, providing mechanistic insights into how alterations in membrane composition, lipid signaling, and energy metabolism may contribute to behavioral and cognitive deficits. Future studies leveraging multi-omics approaches in these models are poised to clarify lipid-centric therapeutic targets for SCZ.
Induced Pluripotent Stem Cell Models of SCZiPSCs have emerged as a powerful platform to model SCZ in a patient-specific context, bridging the gap between clinical observations and mechanistic studies. iPSCs are derived from somatic cells of patients and healthy controls and can be differentiated into relevant neural cell types, including cortical neurons, interneurons, and glial cells [118,156]. This approach allows for the investigation of cellular and molecular phenotypes associated with SCZ, including those related to lipid metabolism.
Recapitulation of Disease-Relevant PhenotypesiPSC-derived neurons from SCZ patients exhibit abnormalities in synaptic connectivity, dendritic spine morphology, mitochondria, and neurotransmitter signaling. Importantly, these cellular phenotypes contribute to the observed defects in neuronal communication and network activity [157–159].
Genetic and Environmental ModelingiPSCs enable the study of both genetic and environmental contributions to SCZ. Patient-derived iPSCs carrying risk alleles in genes such as DISC1, NRG1, SRR, and COMT may show altered lipid metabolic pathways, including phosphatidylinositol and sphingolipid signaling, which can affect synaptic plasticity and neurodevelopment; however, these studies are currently lacking [140,141,160,161]. Additionally, iPSC-derived neurons can be exposed to environmental stressors (e.g., inflammatory cytokines, oxidative stress) to model gene-environment interactions and their impact on lipid homeostasis.
High-Throughput and Multi-Omics ApplicationsiPSC platforms support high-throughput screening and multi-omics approaches, allowing for systematic investigation of lipid-related pathways in SCZ. Lipidomic analyses in iPSC-derived neurons may identify disease-specific lipid signatures and connect them to functional deficits, offering mechanistic insight into how lipid metabolism interacts with synaptic and cognitive abnormalities [162,163].
Unresolved Questions in the Metabolic Foundations of SCZA comprehensive survey and assessment of the literature brings forth the following unanswered questions:
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
-
-
-
-
-
-
-
-
-
-
-
Insights into the molecular mechanisms of lipid dysregulation and altered phospholipid metabolism in SCZ will provide a foundation for novel therapeutic strategies. Understanding how these molecular alterations affect synaptic function, neuronal signaling, and neurodevelopment may guide the development of targeted interventions, including pharmacological agents, dietary modulation, or lifestyle approaches. Molecular profiling could also enable patient stratification, early detection of high-risk individuals, and monitoring of treatment response. Ultimately, integrating these molecular insights with clinical care has the potential to improve symptom management, reduce metabolic comorbidities, and personalize therapy in SCZ.
Biomarkers and Diagnostic PotentialMolecular and metabolic alterations in SCZ, including changes in lipid composition, oxidative stress markers, and synaptic proteins, hold promise as biomarkers for early diagnosis and disease stratification. Lipidomics and metabolomics profiling of peripheral blood or CSF may identify disease-specific signatures, enabling patient-specific risk assessment and monitoring of treatment response. Integration with genetic, imaging, and iPSC-derived cellular data could further enhance diagnostic precision and guide personalized therapeutic strategies.
Future Directions/PerspectivesUnderstanding metabolic and molecular alterations in SCZ is laced with challenges, including patient heterogeneity, variable disease progression, and confounding effects of medication and lifestyle. However, advances in iPSC models, lipidomics, multi-omics integration, and high-resolution imaging offer unprecedented opportunities to uncover disease mechanisms. These tools may enable the identification of novel biomarkers, therapeutic targets, and personalized interventions, bridging the gap between molecular insight and clinical application.
Future research in SCZ will benefit from integrating molecular, metabolic, and cellular insights with clinical data to unravel disease mechanisms and heterogeneity. Advances in iPSC models, organoids, multi-omics, and high-resolution imaging promise to illuminate how lipid and metabolic dysregulation contribute to symptom development. Translating these insights into early diagnostics, targeted therapies, and personalized interventions offers a path toward improved outcomes and precision care for individuals with SCZ.
Conclusions and OutlookSCZ is increasingly recognized as a disorder with profound metabolic and lipid dysregulation that intersects with neurodevelopment, synaptic function, and neurotransmitter signaling. Experimental models, including iPSCs, animal studies, and multi-omics approaches, have begun to uncover the molecular mechanisms linking lipid homeostasis to cognitive and behavioral deficits. Despite challenges such as patient heterogeneity and the influence of medications, these insights offer promising avenues for identifying biomarkers, therapeutic targets, and personalized interventions. Moving forward, integrating molecular, cellular, and clinical data will be essential to translate these discoveries into effective strategies for early diagnosis, treatment, and improved patient outcomes, positioning metabolic and lipid-focused research at the forefront of SCZ therapeutics.
“Not applicable” for studies not involving humans or animals.
Informed Consent Statement “Not applicable.” for studies not involving humans.
Declaration of Helsinki STROBE Reporting GuidelineThis study adhered to the Helsinki Declaration. The Strengthening the Reporting of Observational studies in Epidemiology (STROBE) reporting guideline was followed.
No data were generated from the study.
The author declares no conflicts of interest.
This work was funded by the Maryland Stem Cell Research Grant 2025-MSCRFL-0002 and the University of Maryland, Department of Obstetrics, Gynecology, and Reproductive Sciences Departmental Grant DP255 to RR.
I am deeply grateful to my mentor, Solomon H. Snyder (Sol), at the Johns Hopkins University School of Medicine. As a psychiatrist and pioneering neuroscientist, Sol fostered an environment of intellectual freedom in his lab, allowing me to explore the fascinating world of novel neurotransmitters. It was through the study of D-serine dependent lipid metabolism in serine racemase knockout mice, a model relevant to SCZ, that the inspiration for this review article took shape.
This article is dedicated to the patients, families, and caregivers who live each day with the profound challenges of SCZ. Their strength, resilience, and hope in the face of this lifelong illness continue to inspire and drive the search for understanding and healing.
I sincerely regret that, due to space constraints, I was unable to include all the important work related to this manuscript. I deeply appreciate the contributions of all the researchers in this field and hope this review still honors their invaluable efforts.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
80.
81.
82.
83.
84.
85.
86.
87.
88.
89.
90.
91.
92.
93.
94.
95.
96.
97.
98.
99.
100.
101.
102.
103.
104.
105.
106.
107.
108.
109.
110.
111.
112.
113.
114.
115.
116.
117.
118.
119.
120.
121.
122.
123.
124.
125.
126.
127.
128.
129.
130.
131.
132.
133.
134.
135.
136.
137.
138.
139.
140.
141.
142.
143.
144.
145.
146.
147.
148.
149.
150.
151.
152.
153.
154.
155.
156.
157.
158.
159.
160.
161.
162.
163.
164.
165.
166.
167.
168.
Roychaudhuri R. Altered lipid landscapes provide a molecular perspective on schizophrenia. J Psychiatry Brain Sci. 2025;10(6):e250016. https://doi.org/10.20900/jpbs.20250016.

Copyright © Hapres Co., Ltd. Privacy Policy | Terms and Conditions




