Location:Home >> Detail
J Psychiatry Brain Sci. 2017; 2(5): S7; https://doi.org/10.20900/jpbs.20170014S7

1 Professor of Molecular Psychiatry, Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Center, Nijmegen, The Netherlands.
*Correspondence: Barbara Franke.
Reading the Perspective paper by Boyle, Li, and Pritchard entitled “An expanding view of complex traits: from polygenic to omnigenic”[1], I was torn initially between feelings of doubt and accord. Under a very provocative title, the authors present an excellent integrational analysis of current knowledge, and summarize in clear words what many scholars in the field would ultimately concur with. Putting multifactorial traits and disorders into the framework of an “omnigenic” model seems like an overinterpretation of the evidence at first, but is subsequently qualified by the authors by giving their definition of omnigenicity on page 1182 as “...essentially any gene with regulatory variants in at least one tissue that contributes to disease pathogenesis is likely to have nontrivial effects on risk for that disease.”
In the first part of their analyses, the authors make the point that, when defining trait-/disease-associated alleles based on linkage disequilibrium (LD) around findings from single variant genome-wide association studies (GWAS), most of the genome is associated with the trait/disease. GWAS of height is used as an example, and similar results have been published for schizophrenia previously [2]. While this rather broad distribution of association may suggest an absence of specificity, the authors – like many others cited in their paper – confirm that signal is enriched near genes expressed in disease- / trait-related cell types. However, in two out of three diseases (schizophrenia, rheumatoid arthritis, Crohn disease) used as example, the contribution of risk from active chromatin-related signals comes from broadly expressed genes at least to a similar extent to that from genes uniquely expressed in relevant cell types. I concur with the authors, where they hypothesize that this may be due to small world network structure of signal transduction. Indeed, it has become clear that many proteins are utilized in multiple signalling pathways, where specificity for a certain pathway is achieved through spatial localization / restriction rather than uniqueness of the protein. To my opinion, this principle can provide a good explanation for the broad distribution of findings.
Another point raised by the authors is the apparent difference between genes involved in disease through common and rare variants: taking schizophrenia as an example, they indicate that studies of different types of rare variants, i.e. rare and de novo exonic variants [3,4] as well as rare copy-number variants (CNVs) [5] show consistent enrichment of readily interpretable (synaptic) gene categories, whereas findings from GWAS do not [6]. A bit of cation may be necessary at this point, as the papers cited for the rare variant studies represent the first largerscale exome sequencing studies, and the CNV study cited is a large-scale reanalysis of (partly) previously published data, which might explain the consistency of the findings. New data in preparation right now should be awaited to prove or disprove this point.
Whether or not based on rare variants, the perspective given by Boyle and colleagues on the existence of (a limited number of) “core genes” involved in a disease / trait and (multiple) more peripheral genes influencing its etiology and severity through more diffuse mechanisms nicely complements insights from phenotypic and genetic studies in different psychiatric disorders. For example, working in the field of ADHD, a few recent developments have been important to my understanding of disease etiology: firstly, the fact that – consistent with the phenotypic continuity of ADHD-traits in the general population and clinically diagnosed ADHD – there is also near perfect overlap between the common genetic factors contributing to ADHD and ADHD-traits (rg = 0.94; [7]); secondly, the existence of genes associated with susceptibility to several disorders in GWAS [8-10] and rare variant studies [11] in combination with the existence of a heritable “Psychiatry (P)-factor”, which explains variance across externalizing, internalizing, and autistic population traits [12]; thirdly, the increasing knowledge on the phenotypic and genetic overlap of ADHD with non-brain-centered disorders and traits [7,13,14]. Together, those data provide a picture of a multi-layered genetic contribution to ADHD (but also more generally to a psychiatric disorder), with core genes tightly linked to a (few) disorder(s), a second layer of more general “P-factor” genes, and a third layer of highly general “fitness” genes (e.g. those involved in inflammation or oxidative stress) (Fig. 1). The second and third layer may represent Boyle’s “peripheral genes”. The borders between the three layers are probably gradual.
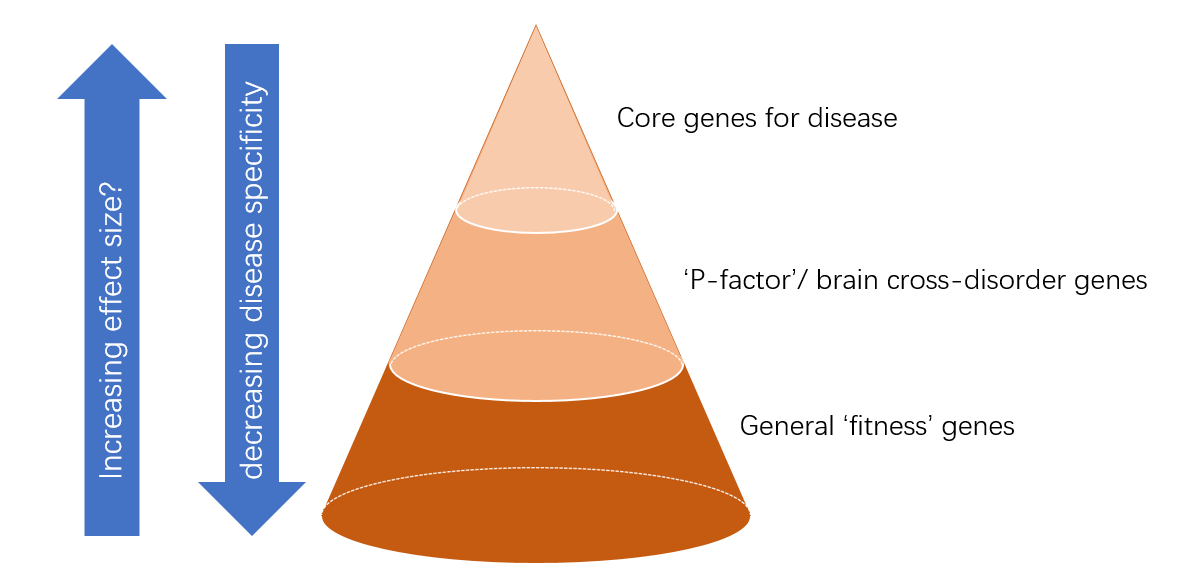 Fig. 1 Model of the hierarchy of genetic contributors to psychiatric disorders, inspired by recent findings
in phenotypic and genetic studies of psychiatric disorders/traits and the omnigenic model presented by
Boyle and colleagues.
Fig. 1 Model of the hierarchy of genetic contributors to psychiatric disorders, inspired by recent findings
in phenotypic and genetic studies of psychiatric disorders/traits and the omnigenic model presented by
Boyle and colleagues.
In summary, the perspective paper by Boyle and coworkers provides a useful point of departure for studies into the genetic architecture underlying human complex diseases and traits. To me, the term “polygenic” seems to provide sufficient room to describe the model put forward by the authors, avoiding the chance of misinterpretation of the term “omnigenic”.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
Franke B. What’s in a Name: the “Omnigenic” Model as a Point of Departure for Polygenic Psychiatric Disorders. J Psychiatry Brain Sci. 2017; 2(5): S7; https://doi.org/10.20900/jpbs.20170014S7

Copyright © 2020 Hapres Co., Ltd. Privacy Policy | Terms and Conditions




