Location: Home >> Detail
Crop Breed Genet Genom. 2026;8(1):e260008. https://doi.org/10.20900/cbgg20260008
 ,
Manoj Kumar Reddy Allam 2 ,
Tulasi Korra 3 ,
Khizar Razzaq 4 ,
Muhammad Massub Tehseen 5 ,
Srushtideep Angidi 4,*
,
Manoj Kumar Reddy Allam 2 ,
Tulasi Korra 3 ,
Khizar Razzaq 4 ,
Muhammad Massub Tehseen 5 ,
Srushtideep Angidi 4,*
1 Department of Plant Science and Landscape Architecture, University of Maryland, College Park, MD 20742, USA
2 Department of Plant and Soil Sciences, Mississippi State University, Starkville, MS 39762, USA
3 Department of Plant Pathology, Banaras Hindu University, Varanasi, UP 221005, India
4 Department of Plant Pathology, North Dakota State University, Fargo, ND 58108-6050, USA
5 Department of Plant Science, North Dakota State University, Fargo, ND 58108-6050, USA
* Correspondence: Srushtideep Angidi
Millets are climate-resilient, nutritionally rich cereal crops that play a critical role in food and nutritional security across arid and semi-arid regions. Despite their inherent tolerance to harsh environments, millet productivity is increasingly challenged by combined biotic and abiotic stresses, including drought, heat, salinity, and emerging diseases. Transcription factors are central regulators of plant stress responses, and among them, WRKY transcription factors constitute one of the largest and most plant-specific families, coordinating defense and stress-adaptive pathways. WRKY transcription factors are characterized by a conserved WRKY domain and zinc finger motif that enable binding to W-box cis-elements in target gene promoters, thereby regulating stress-responsive gene expression. This review synthesizes current knowledge on the structure, classification, and functional roles of WRKY transcription factors in major millet crops, including pearl millet, finger millet, foxtail millet, and related species. We summarize recent advances in millet genomic and transcriptomic resources that have facilitated genome-wide identification of WRKY gene families and their stress-responsive expression patterns. Particular emphasis is placed on WRKY-mediated regulation of disease resistance, abiotic stress tolerance, and hormonal crosstalk involving salicylic acid, jasmonic acid, ethylene, and abscisic acid signaling. Emerging evidence highlights WRKYs as central regulatory hubs that integrate biotic and abiotic stress signals via MAPK cascades, transcription factor networks, and redox signaling. Finally, the potential of WRKY transcription factors to improve millet through molecular breeding, genome editing, and marker-assisted selection is discussed, along with key challenges and future research directions.
Millets, commonly referred to as nutri-cereals, are a diverse group of small-seeded cereal crops that play a crucial role in global food and nutritional security, particularly in developing countries. Major millets such as pearl millet (Pennisetum glaucum), finger millet (Eleusine coracana), foxtail millet (Setaria italica), sorghum, and kodo millet are traditionally cultivated across Asia and Africa, where they serve as staple foods for millions of people living in marginal and rainfed agro-ecosystems [1,2]. Millets are nutritionally superior to many major cereals, being rich in dietary fiber, essential amino acids, minerals such as iron and calcium, and bioactive compounds, while exhibiting low to moderate glycemic indices that help address malnutrition and lifestyle-related diseases [3].
India is the world’s largest producer of millets, accounting for more than 40% of global production, followed by China and Nepal [4]. Although the area under millet cultivation has declined over the past few decades, productivity has steadily increased due to improved agronomic practices and renewed consumer interest. Recognizing their nutritional, environmental, and economic significance, the Food and Agriculture Organization of the United Nations declared 2023 as the International Year of Millets, to promote sustainable millet-based agriculture and enhance global awareness [4].
Millets are widely regarded as climate-smart crops due to their high water-use efficiency, short growth cycles, and adaptability to marginal environments, enabling them to thrive under low-input and climate-variable conditions [5]. However, despite their inherent stress tolerance, millet productivity is increasingly threatened by multiple biotic and abiotic stresses. Abiotic stresses such as drought, heat, salinity, and erratic rainfall patterns severely affect yield stability, particularly in rainfed systems that dominate millet cultivation [6]. Biotic stresses, including fungal diseases (e.g., blast and smut), insect pests (e.g., stem borers and shoot fly), and emerging pathogens, further constrain production due to limited availability of resistant cultivars and inadequate pest and disease management strategies [7,8].
To withstand these challenges, plants rely on complex regulatory networks that integrate stress perception, signal transduction, and transcriptional reprogramming. Transcription factors (TFs) are central components of these networks, acting as molecular switches that regulate stress-responsive gene expression. Among the various TF families, WRKY transcription factors represent one of the largest and most plant-specific groups involved in regulating both biotic and abiotic stress responses [9]. WRKY proteins are characterized by a conserved WRKY domain and a zinc finger motif, which enable them to bind W-box cis-elements in the promoters of target genes and modulate downstream defense pathways [10].
Extensive studies in model plants and major cereals such as Arabidopsis thaliana, rice, maize, and soybean have demonstrated the pivotal role of WRKY transcription factors in pathogen defense, hormonal signaling, reactive oxygen species (ROS) homeostasis, and tolerance to environmental stresses [9,11]. Functional studies of WRKY genes, such as AtWRKY33, OsWRKY45, and GmWRKY54, have demonstrated their involvement in salicylic acid-, jasmonic acid-, ethylene-, and abscisic acid-mediated signaling pathways, as well as in systemic acquired resistance and oxidative stress management [11,12].
In millets, recent advances in next-generation sequencing, transcriptomics, and comparative genomics have enabled the identification of WRKY gene families and stress-responsive expression patterns across species such as pearl millet, foxtail millet, and finger millet [13–15]. However, functional characterization of WRKY transcription factors in millets remains limited, particularly in the context of disease resistance, where most evidence is derived from expression profiling rather than gene-level validation. This gap highlights the need for functional genomics and genome-editing approaches to validate WRKY-centered regulatory networks in millets.
This review synthesizes current knowledge on WRKY transcription factors in major millets, focusing on their structural features, regulatory roles in disease resistance and environmental stress responses, and emerging applications in crop improvement. By integrating structural, regulatory, and functional insights, this review provides a framework for advancing WRKY-based strategies to enhance stress resilience and productivity in millet systems.
WRKY transcription factors (WRKY TFs) represent one of the largest families of plant-specific regulatory proteins and are defined by the presence of a highly conserved WRKY domain of approximately 60 amino acids [9,10]. The hallmark of this domain is the WRKYGQK heptapeptide motif, which mediates binding to W-box cis-elements (TTGACC/T) in the promoters of stress-responsive genes [10]. A schematic representation of the WRKY protein structure, highlighting the WRKY domain, WRKYGQK motif, zinc finger region, and W-box binding, is shown in Figure 1A.
Adjacent to the WRKY motif, WRKY proteins contain a zinc finger-like structure that stabilizes the WRKY domain and facilitates DNA–protein interactions. This zinc finger motif is composed of conserved cysteine (C) and histidine (H) residues that coordinate a zinc ion, ensuring the structural integrity of the DNA-binding domain [16]. Two principal zinc finger types have been identified in WRKY proteins: C2H2 and C2HC, contributing to functional diversification within the family [9,16]. Beyond DNA binding, WRKY transcription factors function within multiprotein regulatory complexes, interacting with other transcription factors, kinases, and chromatin-associated proteins to integrate hormonal and stress-related signals [11].
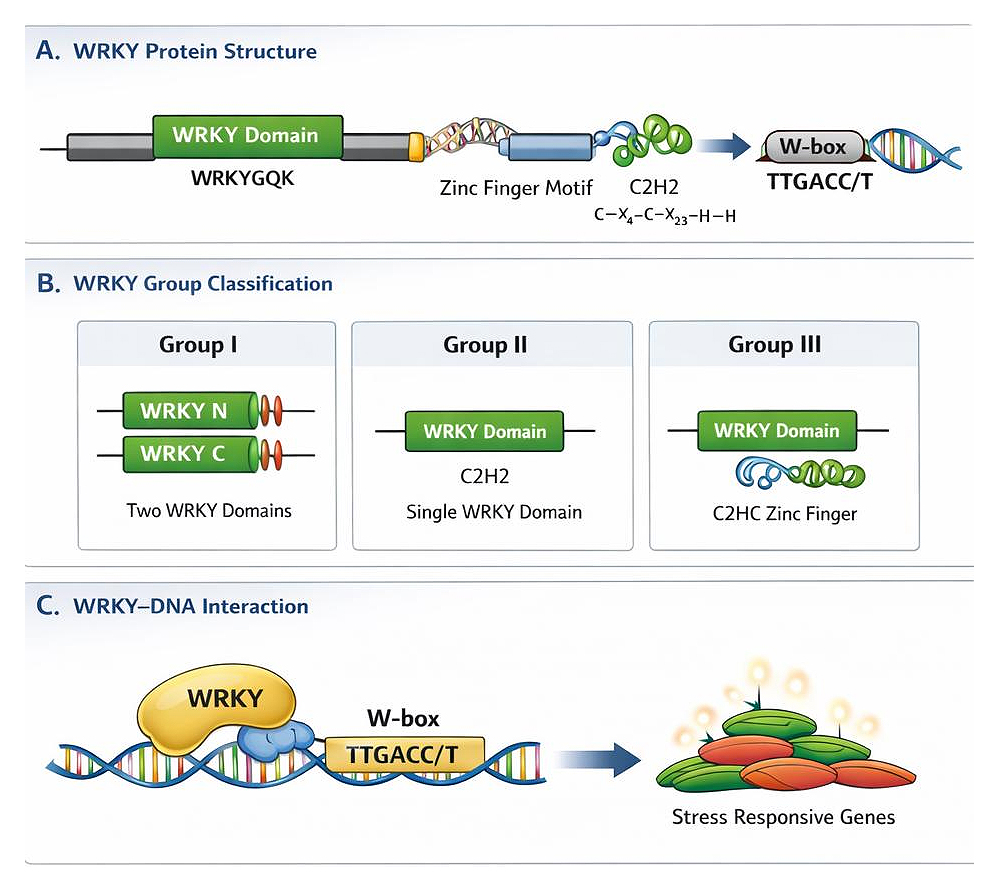 Figure 1. Structure and classification of WRKY transcription factors. (A) Schematic representation of a WRKY protein showing the conserved WRKY domain with the WRKYGQK motif and associated zinc finger motif involved in W-box (TTGACC/T) binding. (B) Classification of WRKY transcription factors into Group I (two WRKY domains with C2H2 zinc finger), Group II (single WRKY domain with C2H2 zinc finger), and Group III (single WRKY domain with C2HC zinc finger). (C) Model illustrating WRKY binding to W-box elements and regulation of stress-responsive gene expression.
Figure 1. Structure and classification of WRKY transcription factors. (A) Schematic representation of a WRKY protein showing the conserved WRKY domain with the WRKYGQK motif and associated zinc finger motif involved in W-box (TTGACC/T) binding. (B) Classification of WRKY transcription factors into Group I (two WRKY domains with C2H2 zinc finger), Group II (single WRKY domain with C2H2 zinc finger), and Group III (single WRKY domain with C2HC zinc finger). (C) Model illustrating WRKY binding to W-box elements and regulation of stress-responsive gene expression.
WRKY transcription factors are classified into three major groups, Group I, Group II, and Group III, based on the number of WRKY domains they contain and the type of zinc finger motif present (Table 1) [10,16]. This classification framework is conserved across plant species, including monocots such as millets, and reflects both structural organization and evolutionary divergence. The structural features distinguishing these groups are illustrated in Figure 1B and Table 1.
Group I WRKY proteins contain two WRKY domains at the N- and C-termini, each harboring the conserved WRKYGQK motif and typically associated with a C2H2-type zinc finger structure [16]. The C-terminal domain generally exhibits stronger DNA-binding affinity, whereas the N-terminal domain is believed to enhance regulatory flexibility and facilitate protein–protein interactions [10]. Group I WRKYs are considered evolutionarily ancestral and are involved in diverse biological processes, including basal defense, developmental regulation, and stress adaptation [9].
Group II WRKY transcription factors possess a single WRKY domain coupled with a C2H2-type zinc finger motif and constitute the largest and most functionally diverse WRKY group. Phylogenetic analyses further subdivide Group II members into five subgroups (IIa–IIe) based on conserved auxiliary motifs and sequence similarity [16]. Group II WRKYs are widely implicated in both abiotic and biotic stress responses, including drought, salinity, oxidative stress, and pathogen defense. Many WRKY genes involved in hormone-mediated signaling and stress-responsive transcriptional regulation belong to this group, highlighting its central role in adaptive stress responses [9,11].
Group III WRKY transcription factors also contain a single WRKY domain but are distinguished by a C2HC-type zinc finger motif, unlike the C2H2 configuration observed in Groups I and II [16]. This structural variation reflects evolutionary divergence and is particularly prominent in monocot lineages. Group III WRKYs are frequently associated with biotic stress responses and are thought to contribute to pathogen recognition, defense gene activation, and stress-induced transcriptional reprogramming. Expansion of Group III members in several grasses, including millets, suggests their potential importance in monocot-specific stress adaptation [9,14].
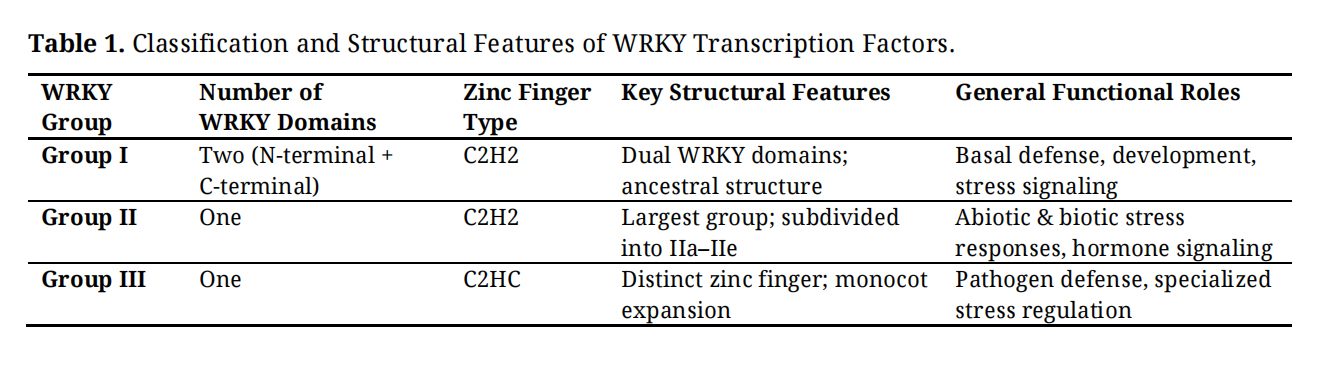 Table 1. Classification and Structural Features of WRKY Transcription Factors.
Table 1. Classification and Structural Features of WRKY Transcription Factors.
The functional versatility of WRKY transcription factors is closely associated with their structural diversity, which arises from variation in WRKY domain number, zinc finger configuration, and conserved sequence motifs. These structural features influence DNA-binding specificity and transcriptional regulatory capacity, enabling WRKY transcription factors to participate in a wide range of stress-responsive pathways. Differences in zinc finger motifs, particularly the C2H2 and C2HC configurations, further contribute to the diversification of regulatory activity among WRKY groups [11,16].
Beyond structural classification, WRKY transcription factors function as key regulatory nodes within plant stress signaling networks. They integrate hormonal signaling pathways and transcriptional crosstalk with other transcription factor families to coordinate appropriate gene expression responses under stress conditions. Through binding to W-box cis-elements in target gene promoters, WRKY proteins regulate the expression of genes involved in defense, stress tolerance, and cellular homeostasis. A simplified conceptual representation of WRKY–DNA interaction and activation of stress-responsive genes is shown in Figure 1C, highlighting the central role of WRKY transcription factors as transcriptional regulators in plant stress adaptation.
Major millets such as pearl millet, finger millet, and foxtail millet are climate-resilient cereals cultivated in arid and semi-arid regions of Asia and Africa. Their adaptability to low rainfall, poor soils, and high temperatures makes them important crops under changing climatic conditions [2,4]. Pearl millet (Pennisetum glaucum) is the most widely cultivated millet and exhibits strong tolerance to drought, heat, and salinity [2,17]. Finger millet (Eleusine coracana) is an allotetraploid species valued for stress resilience and disease resistance [3,13]. Foxtail millet (Setaria italica) is a diploid C4 grass with a compact genome, widely used as a model system for functional genomics and stress biology in grasses [18].
The rapid advancement of next-generation sequencing (NGS) technologies has substantially expanded genomic resources for major millets, enabling genome-wide identification of stress-responsive genes and transcription factor families such as WRKY. Pearl millet was among the first millets to receive a high-quality reference genome (~1.79 Gb), which facilitated the discovery of genes associated with drought tolerance, heat stress adaptation, and disease resistance [19]. More recently, pangenome analyses have revealed extensive structural variation and stress-adaptive loci across diverse pearl millet germplasm, providing valuable resources for comparative genomics and breeding applications [20]. WRKY transcription factor families have been identified and characterized in pearl millet, supported by RNA-seq datasets generated under dehydration and salinity stress [15].
Foxtail millet possesses a compact genome (~515 Mb) and serves as a model panicoid grass for functional genomics [21,22]. Transcriptome analyses under abiotic stresses have enabled identification and characterization of stress-responsive WRKY transcription factors [14,23]. The availability of mapping populations and high-density SNP markers further strengthens its utility for stress biology research. In finger millet, genomic research has progressed with draft genome assemblies and transcriptome datasets that facilitated identification of stress-responsive genes, including WRKY family members [13,24]. RNA-seq analyses of contrasting genotypes and high-throughput marker development (SSR and SNPs) have supported genetic mapping and functional inference [25,26].
Collectively, the availability of reference genomes, transcriptome datasets, and genotyping platforms provides a strong foundation for comparative and functional genomics in millets. These resources enable systematic investigation of WRKY transcription factors and support translation of genomic insights into stress-resilient breeding strategies (Table 2).
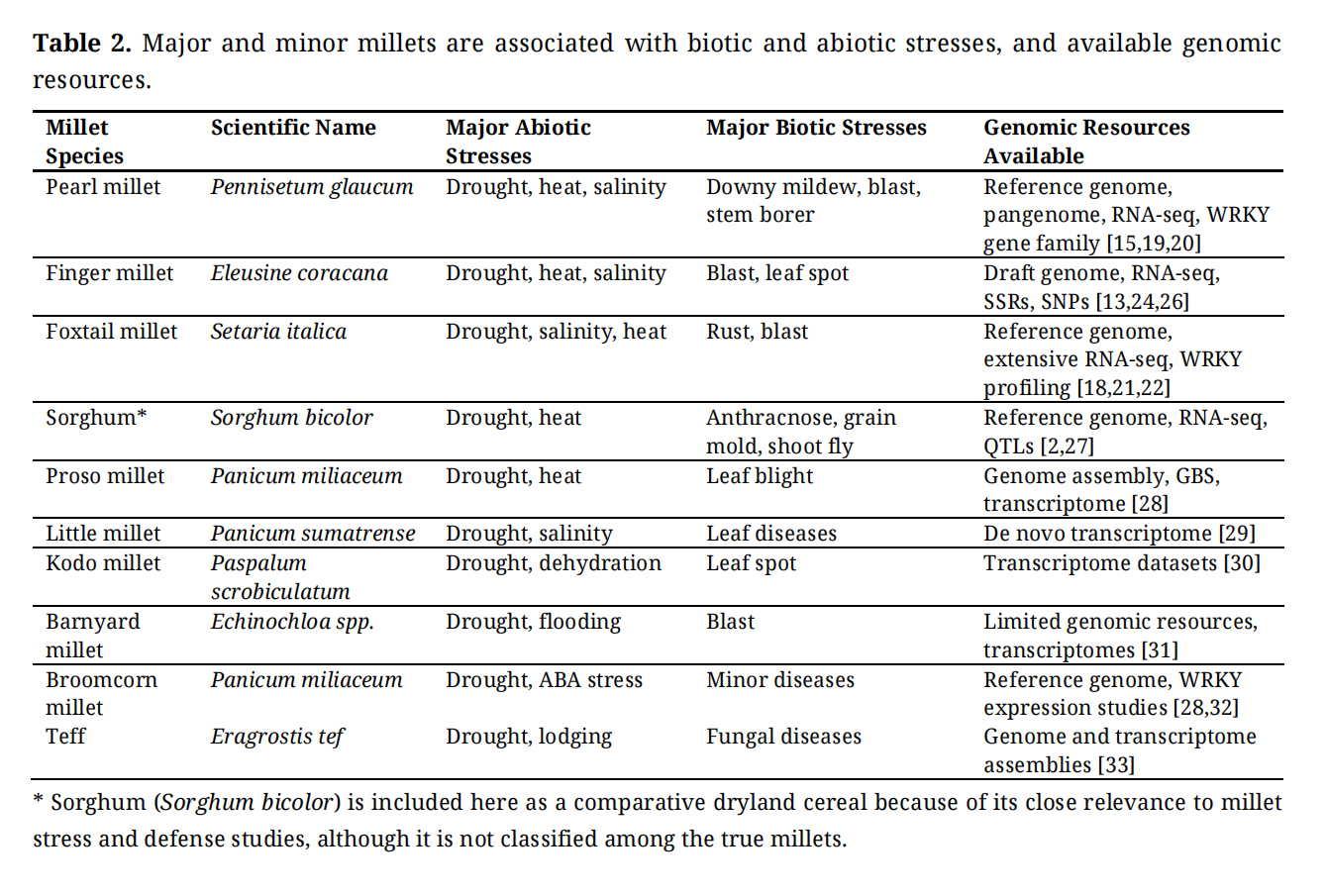 Table 2. Major and minor millets are associated with biotic and abiotic stresses, and available genomic resources.
Table 2. Major and minor millets are associated with biotic and abiotic stresses, and available genomic resources.
The rapid advancement of high-quality reference genomes for major millets has enabled comprehensive evolutionary interrogation of WRKY transcription factor families across monocot grasses. Comparative genomic analyses reveal that although WRKY transcription factors are broadly conserved in structure and classification across land plants, the millet WRKY repertoire displays lineage-specific expansion, structural diversification, and regulatory plasticity, distinguishing monocot cereals from dicot models.
Genome-wide surveys have identified approximately 97 WRKY genes in pearl millet (Pennisetum glaucum), 103 in foxtail millet (Setaria italica), 100 in finger millet (Eleusine coracana), and more than 85 in broomcorn millet (Panicum miliaceum) [13–15]. These values closely align with those of other grasses, such as rice (104) and sorghum (94), yet significantly exceed the 72 WRKY genes reported in Arabidopsis thaliana. The quantitative enrichment of WRKY genes in monocots relative to dicots suggests post-divergence expansion within the grass lineage, potentially associated with ecological adaptation to open, high-radiation, drought-prone environments typical of grassland systems.
Beyond gene counts, the evolutionary architecture of WRKY families in millets reveals multiple layers of complexity. First, subgroup distribution patterns demonstrate a conserved dominance of Group II WRKYs across all millet species, consistent with other monocots [34]. Group II members, particularly subgroups IIc and IId, constitute the largest fraction of the family and are often associated with regulatory flexibility under abiotic stress. However, a striking evolutionary feature in millets is the proportional enrichment of Group III WRKY genes relative to dicot species. In Arabidopsis, Group III members represent a smaller fraction of total WRKYs, whereas in monocot grasses, including pearl millet, foxtail millet, rice, and sorghum, Group III genes are proportionally expanded. This pattern strongly suggests that Group III expansion occurred after monocot–dicot divergence and may reflect adaptive innovation within grass genomes [34].
Phylogenetic reconstruction across monocot WRKY families indicates that Group III genes form multiple grass-specific clades, some of which lack direct orthologs in dicots. These lineage-specific clades likely arose from grass-specific duplication events followed by functional divergence [35]. Group III WRKYs are frequently associated with rapid transcriptional activation during pathogen invasion and stress-induced hormonal signaling. Their expansion may therefore enhance the capacity for rapid transcriptional reprogramming under fluctuating stress regimes, which are common in marginal agroecosystems where millets are cultivated [35].
Gene duplication has played a central role in shaping WRKY family evolution in millets. Multiple mechanisms contribute to WRKY family expansion in millets, including (i) segmental duplications within syntenic chromosomal blocks conserved across panicoid grasses, (ii) tandem duplications generating local WRKY gene clusters on specific chromosomes, and (iii) whole-genome duplication (WGD) and polyploidization events, particularly in finger millet [36].
A comparative collinearity analysis between pearl millet and foxtail millet reveals conserved WRKY loci within syntenic regions, indicating that ancestral duplications preceded panicoid lineage diversification [15]. In finger millet, additional WRKY paralogs likely derive from its polyploid origin, resulting in homoeologous pairs that may exhibit differential expression and regulatory divergence. Polyploidization provides opportunities for neofunctionalization (the acquisition of novel regulatory roles) or sub functionalization (the partitioning of ancestral functions), thereby increasing stress-response specialization. Structural diversification further contributes to WRKY evolutionary complexity. Although WRKY proteins are defined by the conserved WRKYGQK heptapeptide and zinc finger motifs, subtle amino acid substitutions within the WRKY domain have been observed in certain millet paralogs. Variants such as WRKYGKK or WRKYGEK motifs, reported in some grasses, may alter DNA-binding specificity or affinity, potentially enabling diversification of target gene regulation. Additionally, variation in intron–exon structure across WRKY paralogs suggests differential evolutionary pressures and regulatory refinement [32].
Selective pressure analyses in grasses have revealed that while core WRKY domains are generally under purifying selection to preserve DNA-binding function, flanking regions and regulatory domains exhibit signs of diversifying selection. This indicates functional constraint at the structural core combined with adaptive flexibility in regulatory interfaces. Such modular evolution allows WRKY proteins to retain canonical W-box binding while evolving new interaction capabilities with co-regulators such as NAC, MYB, and bZIP transcription factors [36]. Promoter evolution also contributes significantly to WRKY diversification. Comparative cis-regulatory analysis of millet WRKY genes reveals enrichment of stress-related motifs, including W-box, ABRE (ABA-responsive element), MYB-binding sites, and NAC recognition sequences. The combinatorial clustering of these elements suggests that duplicated WRKY genes may acquire differential upstream regulatory control, enabling context-dependent activation under specific stress combinations (e.g., drought–pathogen or salinity–oxidative stress). Thus, evolutionary diversification of regulatory regions may be equally important as coding-sequence divergence in shaping functional specialization [37].
Chromosomal distribution patterns in millet genomes indicate non-random clustering of WRKY genes, with certain chromosomes harboring higher WRKY density. Such clustering often corresponds to duplicated chromosomal segments, reinforcing the role of large-scale duplication events in WRKY expansion. Moreover, lineage-specific retention or loss of particular WRKY paralogs suggests differential adaptive constraints across species [38]. From an adaptive perspective, the expansion and diversification of WRKY transcription factors in millets likely contributed to their remarkable resilience under combined biotic and abiotic stresses. Increased WRKY copy number provides functional redundancy that buffers against loss-of-function mutations and ensures robustness of stress-responsive transcriptional networks. Simultaneously, paralog divergence enables fine-tuned stress discrimination and hormone-specific signaling integration. In particular, the relative enrichment of Group III WRKYs in monocot grasses may enhance pathogen-responsive transcriptional plasticity and rapid activation of defense cascades, traits that are critical in pathogen-rich tropical and subtropical environments [39].
Collectively, comparative evolutionary analysis demonstrates that while WRKY gene families are structurally conserved across land plants, monocot cereals, including millets, exhibit lineage-specific expansion, subgroup enrichment, duplication-driven diversification, and regulatory innovation. These evolutionary patterns provide a molecular basis for the complex stress-adaptive regulatory architecture observed in millet species. Understanding these evolutionary trajectories not only clarifies the functional potential of WRKY transcription factors but also offers a rational framework for prioritizing conserved versus lineage-specific WRKY candidates in stress-resilient millet breeding strategies.
In millet crops, disease-associated WRKY transcription factors have primarily been identified through genome-wide surveys and stress-responsive transcriptome analyses [13,15,32]. Although functional validation remains limited, several WRKYs consistently emerge as strong candidates due to their robust and reproducible induction under pathogen or disease-associated stress conditions (Table 3). These WRKYs are therefore considered central regulators of millet immune responses.
In finger millet (Eleusine coracana), WRKY genes such as EcWRKY29 and EcWRKY33 are strongly induced by blast infection and salinity-associated disease stress, suggesting their involvement in the transcriptional activation of defense pathways [13,25]. Co-expression analyses further indicate that these WRKYs are associated with pathogenesis-related (PR) genes, implying a role in regulating antimicrobial responses during fungal infection [25]. Additionally, EcWRKY45 and EcWRKY70, which are homologs of defense-related WRKYs in other cereals, are upregulated under stress conditions, suggesting potential roles in modulating hormone-dependent immune signaling [40].
In pearl millet (Pennisetum glaucum), several WRKYs stand out as major candidates for disease resistance. PgWRKY33, PgWRKY62, and PgWRKY65 exhibit consistent upregulation under fungal stress and dehydration-linked disease conditions [15,41]. Among these, PgWRKY33 is particularly notable due to its strong stress responsiveness and conserved homology with WRKYs known to regulate defense signaling in monocots [15]. PgWRKY52, although initially characterized for abiotic stress tolerance, also shows transcriptional patterns indicative of crosstalk between abscisic acid (ABA) and jasmonic acid (JA) signaling, highlighting its potential role in integrating stress and immune responses [42].
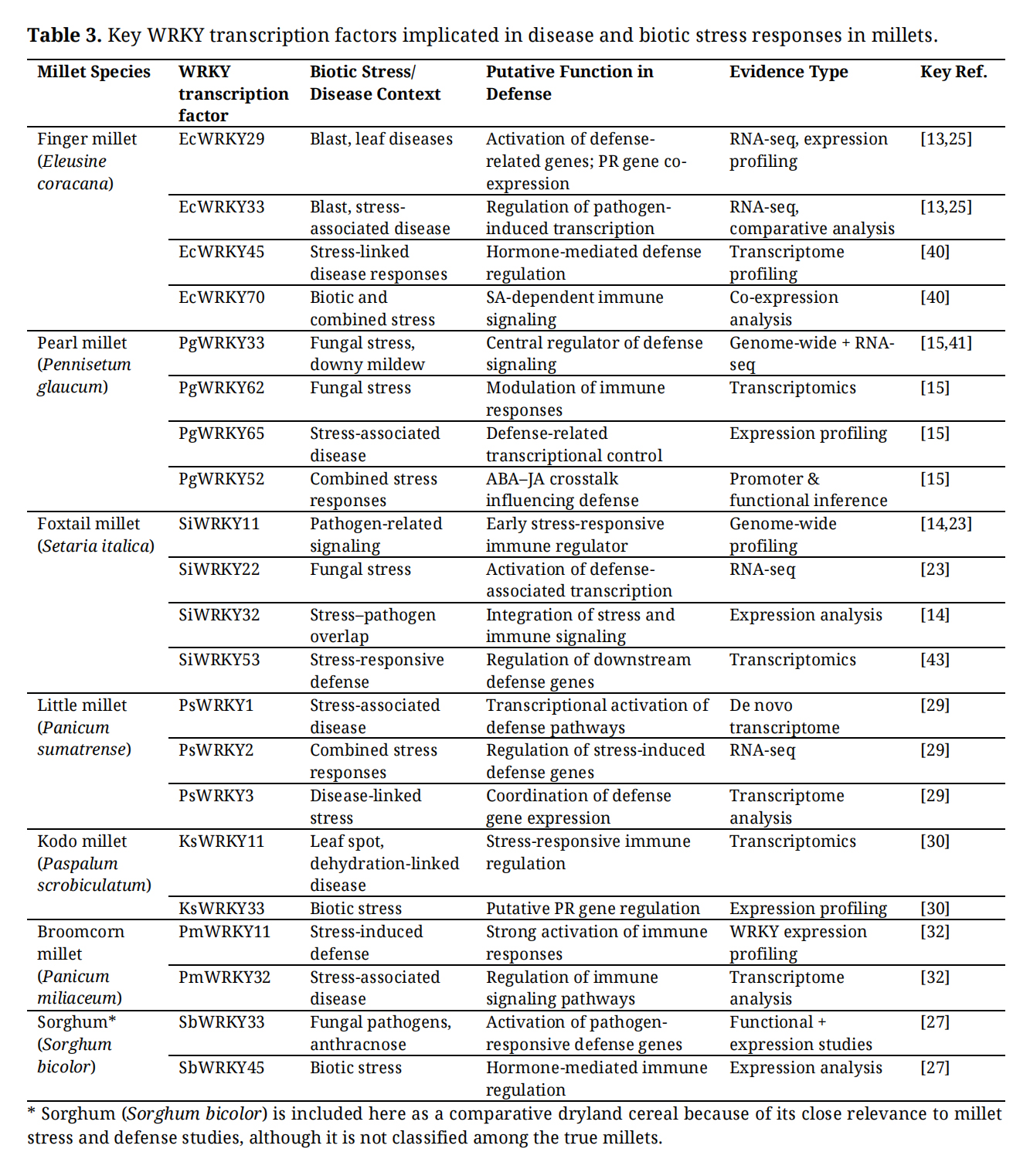 Table 3. Key WRKY transcription factors implicated in disease and biotic stress responses in millets.
Table 3. Key WRKY transcription factors implicated in disease and biotic stress responses in millets.
WRKY transcription factors in millets are closely associated with salicylic acid (SA), jasmonic acid (JA), and ethylene (ET) signaling pathways, which collectively determine resistance outcomes against different classes of pathogens [15,18]. Transcriptomic evidence from pearl millet and finger millet indicates that WRKYs such as PgWRKY33, PgWRKY62, and EcWRKY33 are co-regulated with hormone-responsive defense genes, suggesting their participation in hormonal crosstalk that fine-tunes immune responses [15,25].
In foxtail millet (Setaria italica), genome-wide profiling identified WRKYs including SiWRKY11, SiWRKY22, SiWRKY32, and SiWRKY53 as rapidly induced under stress conditions that overlap with pathogen signaling [18,23,43]. These WRKYs are proposed to function as early-response regulators that activate downstream defense networks, potentially through W-box–mediated regulation of PR gene [14]. The rapid induction of these WRKYs supports their role in priming defense pathways rather than acting as late-stage effectors.
WRKY Regulation of Defense and PR Genes in MilletsThe functional relevance of WRKY transcription factors in millet disease resistance is further supported by promoter analyses and co-expression studies linking WRKYs to PR gene regulation [15,25]. In finger millet and pearl millet, W-box elements are highly enriched in promoters of genes encoding chitinases, β-1,3-glucanases, and defensin-like proteins, consistent with direct WRKY-mediated transcriptional control [13,15]. WRKYs such as EcWRKY29, PgWRKY33, and PgWRKY65 are consistently co-expressed with these PR genes, suggesting direct regulatory involvement during pathogen challenge.
In broomcorn millet (Panicum miliaceum), PmWRKY11 and PmWRKY32 show strong induction under stress conditions associated with defense activation, indicating their involvement in stress-induced immune signaling [32]. Similarly, in little millet (Panicum sumatrense), PsWRKY1, PsWRKY2, and PsWRKY3 are among the most highly expressed transcription factors under combined stress conditions, implying a role in coordinating defense responses during pathogen pressure [29].
Across millet species, WRKYs such as EcWRKY29, EcWRKY33, PgWRKY33, PgWRKY62, PgWRKY65, SiWRKY11, SiWRKY22, and PmWRKY11 consistently emerge as high-confidence candidates due to their strong, stress-specific induction and conserved domain architecture (Table 3) [14,15,32]. Although direct functional validation is still lacking, the convergence of expression-based evidence across species suggests that these WRKYs represent core regulatory nodes in millet immune networks. Targeting these WRKY transcription factors through functional genomics, genome editing, or marker-assisted selection offers a promising strategy for enhancing disease resistance in millet crops.
Millets are widely recognized for their tolerance to drought, heat, and salinity, and WRKY transcription factors constitute a central regulatory component underlying these adaptive responses (Table 4). Genome-wide identification and transcriptome analyses across major and minor millets consistently demonstrate that a substantial proportion of WRKY genes are differentially expressed under abiotic stress conditions, indicating their involvement in stress-responsive transcriptional reprogramming [14,15,29].
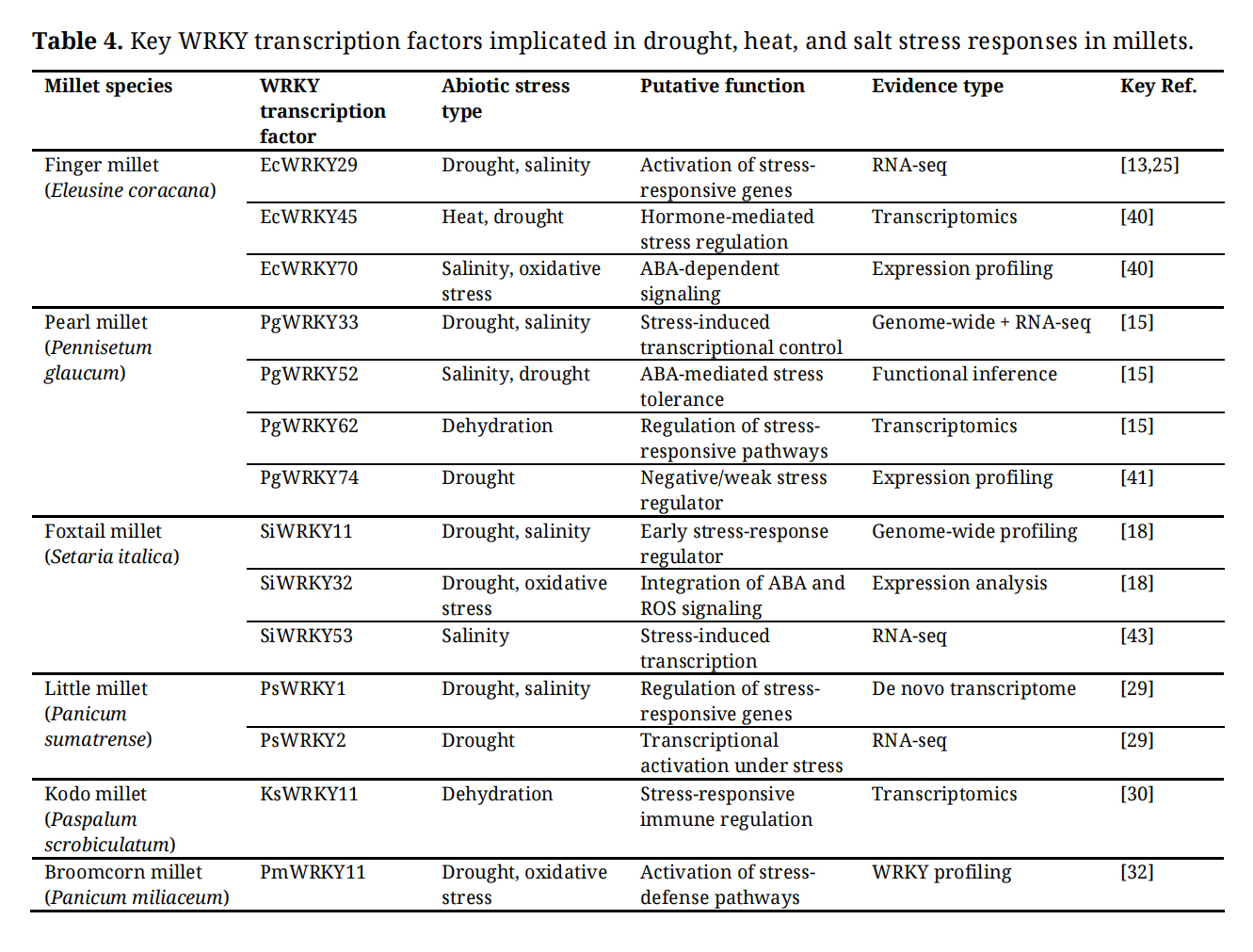 Table 4. Key WRKY transcription factors implicated in drought, heat, and salt stress responses in millets.
Table 4. Key WRKY transcription factors implicated in drought, heat, and salt stress responses in millets.
In finger millet (Eleusine coracana), WRKY transcription factors such as EcWRKY29, EcWRKY45, and EcWRKY70 are strongly induced under drought, heat, and salinity stress, suggesting their role in activating protective stress-response pathways (Table 4) [13,25,40]. Co-expression analyses indicate that these WRKYs are associated with genes involved in osmotic adjustment, antioxidant defense, and stress-related hormonal signaling, highlighting their regulatory role in maintaining cellular homeostasis under adverse environmental conditions [25].
In pearl millet (Pennisetum glaucum), a highly drought- and heat-tolerant C4 cereal, genome-wide WRKY profiling identified PgWRKY33, PgWRKY52, PgWRKY62, and PgWRKY74 as consistently responsive to dehydration and salinity stress [15,41]. Among these, PgWRKY33 and PgWRKY62 exhibit strong transcriptional induction under water-deficit conditions, whereas PgWRKY74 shows weaker or negative regulation, suggesting functional diversification within the WRKY family (Table 4) [15,41].
ABA Signaling and ROS Detoxification Mediated by WRKYsAbscisic acid (ABA) plays a pivotal role in regulating plant responses to drought and salinity, and WRKY transcription factors act as important mediators of ABA-dependent stress signaling in millets. Promoter analyses reveal enrichment of ABA-responsive elements (ABREs) in WRKY-regulated stress-responsive genes, often in combination with W-box motifs, supporting the involvement of WRKYs in ABA signaling cascades [15,29].
In pearl millet, PgWRKY52 has been functionally implicated in enhancing salinity tolerance through ABA-mediated transcriptional regulation, linking WRKY activity with hormonal control of stress adaptation [42]. Similarly, in foxtail millet (Setaria italica), WRKYs such as SiWRKY11, SiWRKY32, and SiWRKY53 are rapidly induced during early drought and salinity stress, suggesting their involvement in ABA-dependent stress priming and early transcriptional activation of downstream protective genes [14,23,43].
WRKY transcription factors also contribute to oxidative stress tolerance by regulating pathways for the detoxification of reactive oxygen species (ROS). Transcriptome studies in finger millet, pearl millet, and little millet report co-expression of WRKYs with genes encoding superoxide dismutase (SOD), catalase (CAT), and peroxidases under drought and salinity stress, indicating a role in maintaining redox homeostasis during prolonged stress exposure [25,29].
Key WRKYs Upregulated under Abiotic Stress Conditions in MilletsComparative analyses across millet species indicate that approximately 20–50 WRKY transcription factors per species are transcriptionally responsive to abiotic stresses such as drought, heat, salinity, and oxidative stress [14,15,29]. However, a smaller subset consistently emerges as high-confidence regulators due to strong, stress-specific induction and conserved WRKY domain architecture.
WRKYs such as EcWRKY29, PgWRKY52, PgWRKY33, SiWRKY11, SiWRKY32, PsWRKY1, and PmWRKY11 represent core components of abiotic stress-responsive transcriptional networks in millets [29,32,42]. Although direct functional validation remains limited for most millet WRKYs, the convergence of expression-based evidence across species underscores their importance as regulatory hubs. Targeting these WRKY transcription factors through genome editing, molecular breeding, or stress-inducible promoter engineering, therefore, represents a promising strategy for enhancing abiotic stress resilience in millet crops.
WRKY transcription factors function as central regulatory hubs embedded within complex signaling networks rather than acting as isolated regulators of stress responses. In millets, accumulating transcriptomic and comparative genomic evidence demonstrates that WRKYs integrate signals from both biotic and abiotic stress pathways, enabling coordinated transcriptional reprogramming under fluctuating environmental conditions [14,15]. This integrative capacity allows millets to balance growth, defense, and stress tolerance in marginal agroecosystems where combined stresses are frequent.
At the upstream level, WRKY activity is closely linked to mitogen-activated protein kinase (MAPK) cascades, which transmit stress perception signals to the nucleus through phosphorylation-dependent activation of transcription factors [44]. Several millet WRKYs responsive to both pathogen challenge and abiotic stress harbor conserved MAPK target motifs, suggesting that post-translational regulation is a key mechanism controlling WRKY function [45]. At the transcriptional level, WRKYs act in coordination with other major transcription factor families, including NAC, MYB, bZIP, and AP2/ERF proteins. NAC transcription factors often function as early stress sensors, while MYBs regulate secondary metabolism, redox balance, and cellular protection. WRKYs fine-tune these responses through W-box–dependent promoter binding, forming co-regulatory modules that have been consistently observed in finger millet, pearl millet, and foxtail millet under drought, salinity, heat, and pathogen stress [25,29].
A defining feature of WRKY-centered networks is their role in integrating defense-related and abiotic stress signaling pathways. WRKY transcription factors serve as convergence points for hormone-mediated signaling involving salicylic acid (SA), jasmonic acid (JA), ethylene (ET), and abscisic acid (ABA), allowing stress responses to be prioritized according to environmental context [11]. Several WRKYs initially identified in disease resistance studies are also transcriptionally activated under drought, salinity, heat, or oxidative stress, highlighting their functional plasticity. WRKYs such as EcWRKY29, EcWRKY33, PgWRKY33, PgWRKY52, and SiWRKY11 exemplify this dual functionality, regulating pathogen-responsive genes during biotic stress while also participating in ABA-mediated stress signaling and reactive oxygen species (ROS) detoxification under abiotic stress [15,32,42]. Rather than acting redundantly, these WRKYs display context-dependent regulatory behavior, activating distinct downstream gene sets depending on stress type, intensity, and hormonal balance.
WRKY regulatory networks are further stabilized by feedback and feedforward loops, including auto-regulation and cross-regulation among WRKY genes themselves. WRKYs frequently bind W-box elements within their own promoters or those of other WRKY genes, forming hierarchical regulatory circuits that amplify early stress responses and facilitate signal attenuation once stress subsides. In addition, WRKY-mediated regulation is tightly coupled with ROS homeostasis, where WRKYs control antioxidant and detoxification genes, while ROS levels reciprocally influence WRKY expression and activity [45]. A conceptual model illustrating MAPK–WRKY interactions, transcription factor crosstalk, hormonal integration, and feedback regulation in millets is presented in Figure 2.
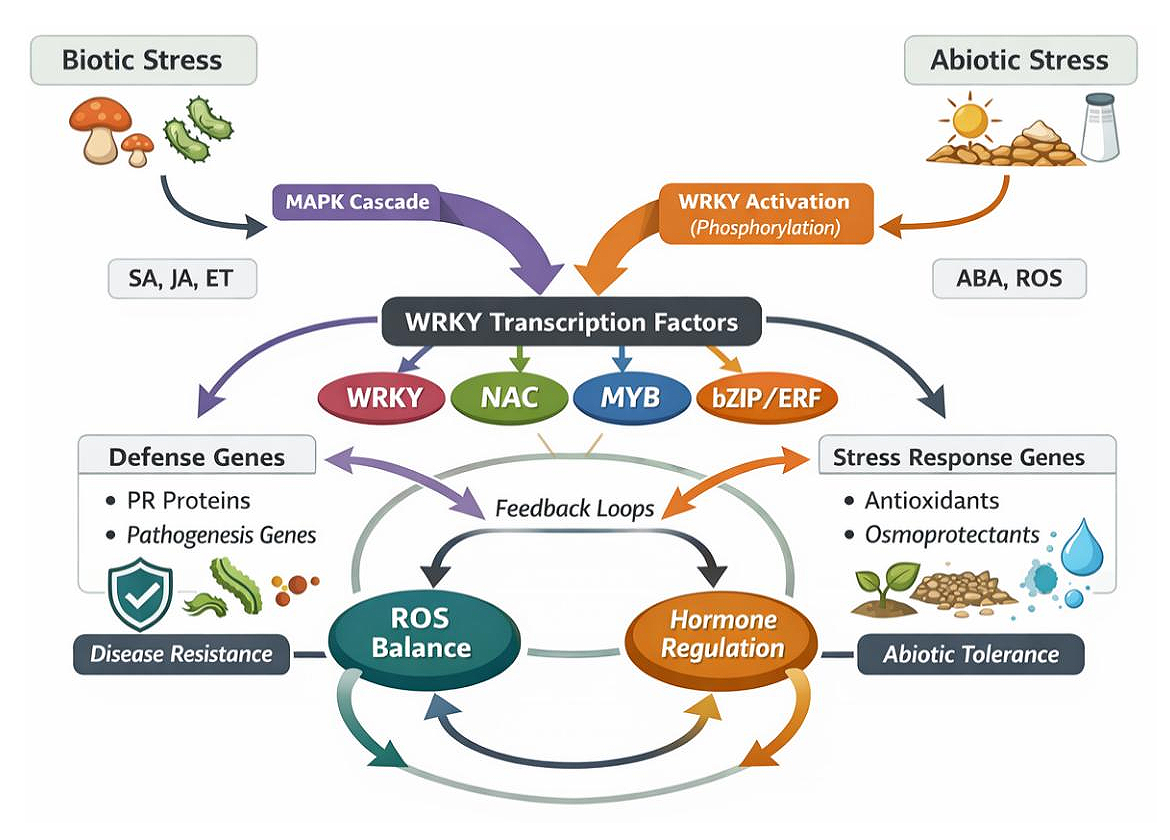 Figure 2. WRKY-centered regulatory networks integrating biotic and abiotic stress signaling in millets Conceptual illustration showing MAPK-mediated activation of WRKY transcription factors, their interaction with NAC, MYB, bZIP, and AP2/ERF transcription factors, integration of SA, JA, ET, and ABA signaling, regulation of defense and stress-tolerance genes via W-box elements, and feedback loops involving ROS homeostasis.
Figure 2. WRKY-centered regulatory networks integrating biotic and abiotic stress signaling in millets Conceptual illustration showing MAPK-mediated activation of WRKY transcription factors, their interaction with NAC, MYB, bZIP, and AP2/ERF transcription factors, integration of SA, JA, ET, and ABA signaling, regulation of defense and stress-tolerance genes via W-box elements, and feedback loops involving ROS homeostasis.
Promoter and motif analyses have provided important insights into the transcriptional regulatory mechanisms underlying WRKY-mediated stress responses in millets. Genome-wide surveys and transcriptome-based studies in pearl millet, foxtail millet, finger millet, and other minor millets consistently report a high enrichment of W-box cis-regulatory elements (TTGACC/T) in the promoters of stress-responsive genes, including pathogenesis-related (PR) genes, antioxidant enzymes, and hormone-responsive regulators [14,15,32]. In silico promoter analyses further reveal the co-occurrence of W-box motifs with ABA-responsive elements (ABREs), MYB-binding sites, and NAC recognition motifs, supporting the coordinated action of WRKYs with other transcription factor families during stress adaptation. In millets, such combinatorial promoter architectures are particularly prominent in genes induced under drought–pathogen and salinityoxidative stress conditions, suggesting that WRKYs contribute to context-dependent transcriptional regulation through motif clustering rather than single-element control. Although experimental validation of WRKY–promoter interactions remain limited in millets, these conserved motif patterns strongly support a central role for WRKY transcription factors in integrating hormonal and stress-derived signals at the promoter level.
Overall, the WRKY-centered regulatory architecture characterized by MAPK signaling integration, transcription factor cooperation, hormonal crosstalk, and feedback regulation provides a robust and flexible framework for stress resilience in millets. Understanding these interconnected regulatory modules is critical for identifying key regulatory nodes that can be targeted through functional genomics, genome editing, or marker-assisted breeding to enhance multi-stress tolerance in millet crops.
Mechanistic Basis of Stress-Specific WRKY Responses in MilletsBuilding upon the regulatory framework described above, stress specificity emerges through context-dependent modulation of WRKY activity. Although WRKY transcription factors are frequently described as integrators of both biotic and abiotic stress responses, an important mechanistic question arises: how does a single WRKY protein differentiate between distinct stress types, such as pathogen infection and drought? Stress specificity does not arise from WRKY proteins acting in isolation, but rather from context-dependent modulation involving upstream signaling dominance, promoter architecture, co-factor recruitment, and post-translational modifications.
In response to fungal infection, stress perception via pattern-recognition receptors (PRRs) activates pathogen-responsive MAPK cascades and promotes the accumulation of salicylic acid (SA) or jasmonic acid/ethylene (JA/ET), depending on the pathogen lifestyle. In this context, WRKY proteins, such as PgWRKY33, are phosphorylated by pathogen-activated MAPKs, thereby enhancing their transcriptional activity toward defense-related targets. SA-dominant signaling prioritizes the activation of pathogenesis-related (PR) genes, antimicrobial peptides, and genes involved in secondary metabolite biosynthesis. Promoters of these defense genes typically contain clustered W-box motifs, which are enriched in SA-responsive regulatory elements. Additionally, WRKY proteins may preferentially interact with MYB transcription factors involved in phenylpropanoid and phytoalexin biosynthesis during pathogen stress, reinforcing defense-specific transcriptional programs [46,47].
In contrast, during drought stress, abscisic acid (ABA) becomes the dominant hormonal signal. ABA-mediated signaling activates distinct kinase pathways and alters WRKY activity in a cellular redox environment. Under drought, WRKY proteins often bind promoters containing combinatorial motif architectures, including W-box elements adjacent to ABA-responsive elements (ABREs). This W-box–ABRE coupling facilitates cooperative regulation between WRKYs and bZIP transcription factors, which are primary mediators of ABA signaling. Rather than activating PR genes, WRKY targets under drought typically include genes associated with osmotic adjustment, late embryogenesis abundant (LEA) proteins, antioxidant enzymes, and ROS detoxification pathways [48,49].
Using PgWRKY33 as an illustrative example, stress-specific behavior can be explained by three key mechanistic layers: First, differential promoter binding: although PgWRKY33 recognizes the same W-box core motif, promoter context differs between pathogen-responsive and drought-responsive genes. Motif clustering and neighboring cis-elements influence binding affinity and transcriptional outcome [50]. Second, co-factor interaction differences: under pathogen stress, PgWRKY33 may preferentially interact with MYB transcription factors or defense-related co-activators, whereas under drought conditions, it may interact with NAC or bZIP proteins that modulate ABA-responsive gene expression. These alternative protein–protein interactions shift transcriptional target specificity. Third, post-translational modifications: pathogen-triggered MAPKs may phosphorylate PgWRKY33 at distinct residues compared to ABA-activated kinases. Differential phosphorylation patterns can alter DNA-binding affinity, protein stability, nuclear localization, or interaction with transcriptional co-regulators, thereby influencing downstream gene selection [48,50].
Additionally, cellular redox status contributes to stress discrimination. Pathogen infection often induces a rapid oxidative burst, whereas drought leads to gradual ROS accumulation. WRKY proteins function within this redox context, modulating antioxidant gene expression while being themselves responsive to ROS levels. Thus, ROS dynamics further refine stress-specific transcriptional responses. Taken together, stress specificity of WRKY-mediated regulation arises from integration of hormonal dominance (SA vs ABA), promoter motif architecture (W-box with defense elements vs W-box with ABRE), co-factor recruitment (MYB/NAC/bZIP combinations), and stress-specific post-translational modifications. Rather than functioning as binary switches, WRKY transcription factors act as modular regulatory hubs whose output is shaped by cellular signaling context [51,52].
Functional Validation of WRKY Genes: Evidence from Model Systems and Implications for MilletsAlthough transcriptomic and genome-wide analyses have identified numerous stress-responsive WRKY transcription factors in millets, direct functional validation of individual genes remains limited. Nevertheless, extensive experimental evidence from model plants and major cereals provides strong support for conserved WRKY-mediated regulatory mechanisms in biotic and abiotic stress responses, offering a robust framework for functional inference in millet species.
In Arabidopsis thaliana, AtWRKY33 is among the most extensively characterized WRKY regulators. Loss-of-function mutants of AtWRKY33 exhibit pronounced susceptibility to the necrotrophic fungus Botrytis cinerea, accompanied by reduced camalexin biosynthesis and impaired activation of defense-associated genes. Mechanistically, AtWRKY33 functions downstream of MAPK cascades and activates jasmonic acid (JA)- and ethylene (ET)-dependent defense pathways by directly binding W-box elements in target promoters [53,54]. These studies demonstrate that WRKY33 acts as a critical transcriptional node linking pathogen perception to antimicrobial gene expression.
In monocot cereals, OsWRKY45 in rice provides compelling evidence of WRKY-mediated immune regulation. Overexpression of OsWRKY45 enhances resistance to rice blast (Magnaporthe oryzae) and bacterial leaf blight by activating salicylic acid (SA)-dependent signaling and inducing pathogenesis-related (PR) genes. Importantly, OsWRKY45-mediated resistance has been validated under field conditions, indicating that WRKY-based genetic manipulation can confer durable disease resistance when expression is appropriately regulated [55,56]. These findings underscore the translational potential of WRKY genes in cereal crop improvement.
Similarly, SbWRKY33 in sorghum has been implicated in fungal defense responses, with strong induction following pathogen challenge. Functional analyses suggest conservation of MAPK–WRKY–PR regulatory modules across grasses. In soybean, GmWRKY54 has been shown to regulate reactive oxygen species (ROS) homeostasis under stress conditions [12,57]. Overexpression lines exhibit enhanced tolerance associated with increased expression of antioxidant enzymes such as superoxide dismutase (SOD) and catalase (CAT), highlighting the role of WRKY transcription factors in redox balance and oxidative stress mitigation.
Collectively, these validated WRKY genes across dicots and monocots reveal conserved mechanistic themes, including MAPK-mediated phosphorylation and activation. Hormonal integration among SA, JA, ET, and ABA pathways. Direct transcriptional regulation via W-box cis-elements. Modulation of ROS detoxification and stress-responsive gene networks. Although functional validation in millets remains at an early stage, homologous evidence strongly supports conserved defense and stress-regulatory roles for millet WRKY orthologs. For instance, PgWRKY33 in pearl millet [15], EcWRKY29 and EcWRKY33 in finger millet [13,25], and SiWRKY11 in foxtail millet [14] display strong stress-inducible expression and high sequence similarity to experimentally validated WRKY regulators in other species. These parallels suggest that millet WRKYs likely operate within conserved MAPK–WRKY–PR and ABA–WRKY–ROS regulatory modules.
To experimentally confirm these roles in millets, targeted functional approaches are needed. CRISPR/Cas-mediated knockout of candidate genes such as PgWRKY33 would enable direct assessment of susceptibility phenotypes under fungal infection or combined drought–pathogen stress conditions. Complementary gain-of-function analyses using stress-inducible promoters could evaluate enhanced resistance while minimizing potential growth penalties associated with constitutive overexpression. Foxtail millet, with its relatively efficient transformation systems and compact genome, provides a promising platform for generating stable overexpression or gene-edited WRKY lines. Additionally, promoter editing strategies targeting W-box or ABA-responsive elements (ABREs) may allow fine-tuning of stress-responsive transcription without altering coding sequences. Integration of gene editing with transcriptomic profiling, hormone quantification, and ROS measurements will be essential to dissect WRKY-centered regulatory circuits in millet systems. Such functional validation will not only confirm conservation of WRKY-mediated stress integration but may also reveal millet-specific regulatory adaptations shaped by evolutionary divergence.
In summary, while direct genetic evidence in millets is still emerging, extensive validation from Arabidopsis, rice, soybean, and sorghum strongly supports the central role of WRKY transcription factors in coordinating immune regulation and abiotic stress tolerance. Translating this mechanistic knowledge into millet species through genome editing and controlled expression strategies represents a critical step toward deploying WRKY-based interventions for multi-stress resilience.
WRKY transcription factors represent promising molecular targets for improving stress resilience in millets due to their central roles in regulating both biotic and abiotic stress responses as shown in Figure 3. Genome-wide identification and expression profiling studies in pearl millet, finger millet, and foxtail millet have revealed several WRKY genes that are consistently induced under drought, salinity, heat, and pathogen challenge, making them attractive candidates for crop improvement programs [14,15,20].
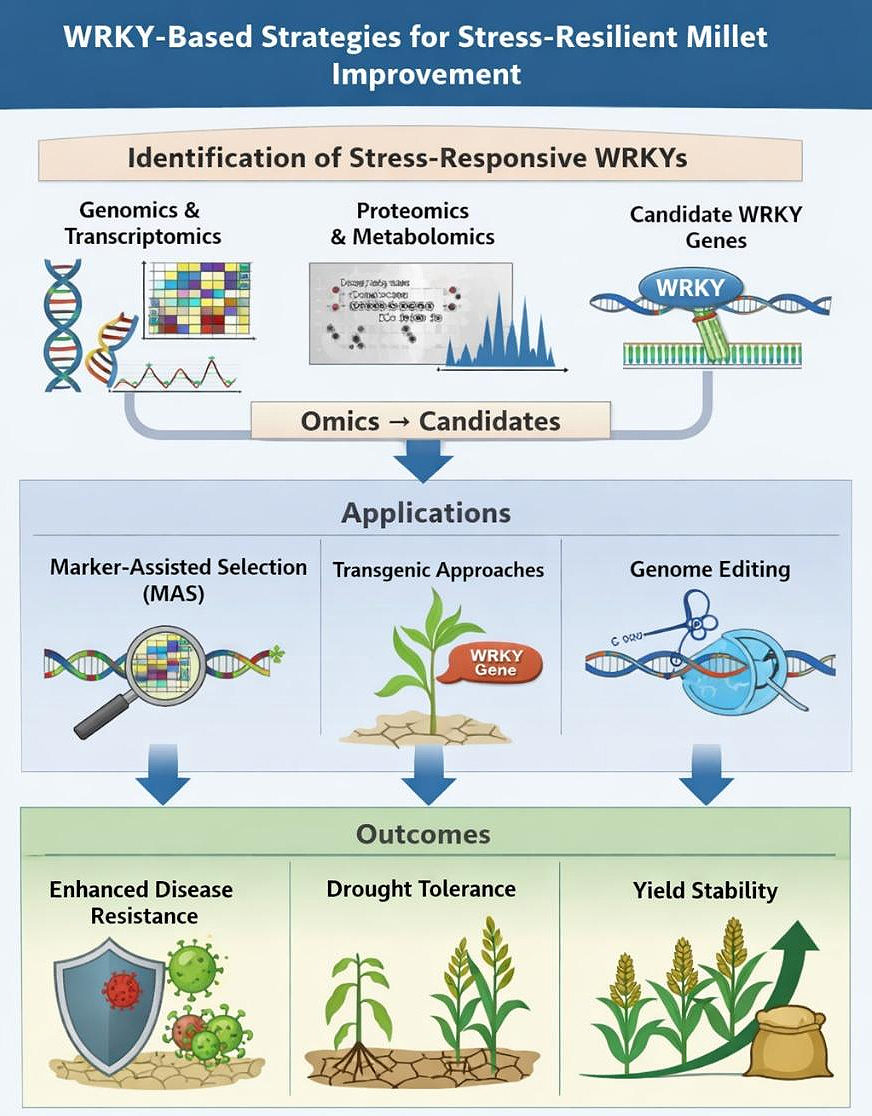 Figure 3. WRKY-based strategies for stress-resilient millet improvement. Conceptual overview showing identification of stress-responsive WRKY transcription factors through omics approaches and their application in marker-assisted selection, transgenics, and genome editing to enhance disease resistance, abiotic stress tolerance, and yield stability in millets.
Figure 3. WRKY-based strategies for stress-resilient millet improvement. Conceptual overview showing identification of stress-responsive WRKY transcription factors through omics approaches and their application in marker-assisted selection, transgenics, and genome editing to enhance disease resistance, abiotic stress tolerance, and yield stability in millets.
WRKY genes with stable stress-responsive expression patterns can be exploited through transgenic or cisgenic strategies to enhance tolerance without compromising yield. In cereals and model plants, overexpression of WRKY homologs has resulted in enhanced disease resistance and improved tolerance to drought and oxidative stress, supporting their functional conservation across grasses. In millets, candidate WRKYs such as EcWRKY33, PgWRKY33, and SiWRKY11 show strong inducibility under multiple stresses, suggesting their suitability for functional validation and transgenic deployment [14,15]. Importantly, stress-inducible or tissue-specific promoters may mitigate potential growth penalties associated with constitutive WRKY expression.
WRKY genes and WRKY-linked genomic regions also hold potential as molecular markers in breeding programs. Stress-associated WRKY loci identified through transcriptome profiling and comparative genomics can be integrated into marker-assisted selection (MAS) pipelines. Given the availability of reference genomes and high-density SNP datasets for several millets, WRKY-anchored markers may facilitate the selection of genotypes with enhanced stress adaptability, particularly in marginal environments [19,24].
The emergence of genome editing technologies such as CRISPR/Cas systems offers new opportunities to fine-tune WRKY-mediated regulatory networks. Targeted editing of WRKY genes or their promoter regions could enhance stress responsiveness while preserving developmental balance. Additionally, stacking WRKYs with complementary functions such as combining defense-related WRKYs with ABA-responsive WRKYs may provide durable tolerance to combined stresses frequently encountered in millet-growing regions [20,28].
Despite significant advances in the identification of WRKY genes, functional characterization in millets remains limited. Most studies rely on transcriptomic induction patterns rather than direct gene validation, creating a gap between candidate discovery and practical application. The lack of stable transformation systems and limited mutant resources in several millet species further constrain functional genomics efforts [14,15].
Future research must adopt system-level approaches, integrating genomics, transcriptomics, epigenomics, and metabolomics to unravel WRKY-centered regulatory networks under single and combined stress conditions. Comparative analyses across millet species and related cereals will be essential to identify conserved WRKY modules with broad applicability. Additionally, expanding functional validation through genome editing and stress-specific phenotyping will be critical for translating WRKY knowledge into resilient millet cultivars.
Ultimately, WRKY transcription factors represent a powerful yet underutilized regulatory layer in millet stress biology. Strategic integration of WRKY-based markers, regulatory networks, and gene-editing technologies holds substantial promise for developing climate-resilient millet varieties suited to future agricultural challenges.
No new data were generated or analyzed in this study. All information presented in this review is based on previously published literature, which is appropriately cited.
SA designed and conceptualized the review and developed the central idea. KR and MKRA contributed to data collection and curation. UKT, TK, MMT, MKRA, SA, and KR contributed to the writing and revision of the manuscript. All authors reviewed and approved the final manuscript.
The authors declare that there is no conflict of interest regarding the publication of this review. Generative artificial intelligence–assisted tools were used to support language refinement and the conceptual design of schematic figures. All scientific content, figure interpretation, and conclusions were critically reviewed, revised, and approved by the authors.
This work did not receive any specific grant from funding agencies in the public, commercial, or non-profit sectors.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
Thera UK, Allam MKR, Korra T, Razzaq K, Tehseen MM, Angidi S. WRKY Transcription Factors in Disease Resistance and Environmental Stress Responses in Millets). Crop Breed Genet Genom. 2026;8(1):e260008. https://doi.org/10.20900/cbgg20260008.

Copyright © Hapres Co., Ltd. Privacy Policy | Terms and Conditions




