Location: Home >> Detail
Immunometabolism. 2021;3(3):e210023. https://doi.org/10.20900/immunometab20210023

Department of Pharmacology and Therapeutics, School of Medicine, Trinity College Dublin, Dublin 2, Ireland
* Correspondence: Margaret Lucitt.
This article belongs to the Virtual Special Issue "Cardiovascular Immunometabolism"
Atherosclerosis is well recognised as a disease associated with elevated cholesterol levels. Innate monocytes and macrophage cells laden with cholesterol have long been described as key players in driving low grade inflammation characteristic of atherosclerosis. In more recent times it has been shown how various mechanisms controlling metabolic and epigenetic reprogramming of these innate immune cells influence their inflammatory responses. In this review a general role of intracellular metabolism in reprogramming innate immune cells will be discussed with a particular emphasis on evidence supporting how innate reprogramming contributes to the pathophysiology of atherosclerosis. In addition the evidence for the role of statins in altering these metabolic adaptations to control the development and progression of atherosclerotic plaques is discussed.
Atherosclerosis is a chronic lipid-driven inflammatory disease of the arterial vasculature and the main underlying pathological cause of cardiovascular disease [1]. During atherosclerosis development, increased circulating levels of low-density-lipoprotein cholesterol (LDL-C) is linked mechanistically and genetically to an increased risk of cardiovascular clinical events [2]. It is now established that atherosclerotic disease progression is also strongly associated with inflammatory processes involving cells of the innate immune system, primarily monocyte-derived macrophages [3,4]. Cholesterol rich lipoproteins provides an immune stimulus as they accumulate in the arterial wall, where they undergo oxidation driving recruitment and polarization of macrophage cells [5]. Targeting hyperlipidaemia in patients has proven very successful in the management of atherosclerosis using pharmacological approaches. Well established agents such as the statins (3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors) inhibit cholesterol synthesis and significantly reduce circulating levels of LDL-C [6]. More recently bempedoic acid [7] has been described to inhibit cholesterol synthesis upstream of statins. Other novel approaches include proprotein convertase subtilisin–kexin type 9 inhibitors (PCSK9i) which reduce LDL receptor turnover and effectively lower LDL-C [8]. Furthermore, approaches to inhibit the synthesis of lipoproteins have also been described, including lomitapide which blocks microsomal triglyceride transfer protein and mipomersen, an antisense oligonucleotide directed against apoB100 [8]. These two latter approaches are particularly useful when there is no or little residual LDL-receptor function as in patients with homozygous familial hypercholesterolaemia. In addition to inhibiting cholesterol synthesis, statins have also been described to play direct roles in modulating innate inflammatory responses, expanding their role in limiting atherosclerosis progression [9].
Macrophages are essential components of tissues throughout the body and act to maintain tissue homeostasis and function as sentinel immune cells [10]. Macrophages infiltrate and reside in healthy tissue, where they play a critical role in maintaining tissue homeostasis during aseptic conditions [11]. These innate cells carry great plasticity and can change their functional phenotype in response to localised tissue environments during pathogenic challenge or encounters with sterile inflammatory mediators during atherosclerosis. Subendothelial accumulation of cholesterol-rich lipoproteins, particularly LDL, at susceptible sites of the arterial wall, triggers the infiltration of innate immune cells particularly macrophages [12,13]. In the vascular intima, LDL particles undergo oxidative modifications to oxidised (Ox)LDL which are engulfed by macrophages to form the characteristic foam cell. (Ox)LDL also leads to further macrophage activation and recruitment [14]. Additionally, T cell recuitment and activation contributes to the atherosclerotic plaque inflammatory component [15]. Ongoing innate and adaptive immune processes promote plaque progression, remodelling and contribute to rupturing of unstable plaques leading to thrombus formation and acute clinical manifestations such as myocardial infarctions and stroke [16].
Similar to other immune cell subsets, cellular metabolism is emerging as a key driver of macrophage function [17]. Intracellular rewiring of metabolic pathways and metabolic intermediates facilitate macrophage responses to extracellular factors such as hyperlipidaemia, oxidative stress, hypoxia, as well as exposure to cellular components from apoptotic and necrotic cell damage. The atherosclerotic plaque is a rich source of these factors and provides an environment that likely influences macrophage responses and their intracellular metabolic wiring [3]. Aerobic glycolysis has been identified as one of the earlier metabolic shifts allowing macrophage switching from a resting to a more activated state of phagocytosis, proliferation and cytokine production [18,19]. Furthermore, recognised changes in other metabolic processes such as amino acid [19] and lipid [20,21] biosynthesis pathways also act to reprogramme macrophages to adapt specific functional responses. Targeting intracellular metabolic pathways in immune cells is evolving as a promising approach to prevent and dampen inflammatory responses, offering significant potential across several inflammatory disease states [22].
This review will discuss in general the role of cellular metabolism in macrophage function and how local and systemic factors in the atherosclerotic plaque have been shown to regulate macrophage cellular metabolism. With an emphasis on how cellular cholesterol lipid metabolism contributes to a proinflammatory state in atherosclerotic disease development and progression we describe accumulating evidence for the role of statins in modulating these events to inhibit disease pathogenesis.
Macrophages become activated by various tissue environmental stimuli leading to a range of cellular functional states. An M1-like state is characterized by the release of proinflammatory cytokines such as tumour necrosis factor-alpha (TNF-α), interleukin (IL) 1β (IL-1β), IL-6, IL-12, and IL-23), whereas an M2-like or alternative state participates in tissue repair and remodelling producing pro-resolving molecules, such as IL-10 and transforming growth factor-β (TGF-β) [23]. The M1/M2 classification, while still in use, is an over-simplified view, based on in vitro models of the now recognised complex phenomena of macrophage polarization in vivo [24]. In addition to such acute polarized activation states, it is now also evident that macrophages, and indeed other cell types, can undergo more sustained long-term functional reprogramming in a process termed trained immunity [25].
Trained immunity in macrophage cells is mediated by epigenetic reprogramming of transcriptional pathways, often during early myeloid progenitor development, which contribute to enhanced effector functions, phenotypes and activation states [26,27]. Such a response has been extensively described as a result of exogenous insults occurring during pathogenic infection, with the fungal cell wall component β-glucan,[28], bacterial cell wall component lipopolysaccharide (LPS) [29] and the Bacillus Calmette–Guérin (BCG) vaccine [30]. These long-term adaptive changes in functional responsiveness are also associated with changes in several cellular metabolic pathways such as glycolysis, tricarboxylic acid (TCA) cycle, amino acid metabolism, fatty acid synthesis and cholesterol metabolism [31]. In particular, a shift from oxidative phosphorylation to aerobic glycolysis (known as the Warburg effect), regulated via a mammalian target of rapamycin (mTOR) and hypoxia-inducible factor-1 α (HIF-1α) signalling pathway, is a central mechanistic component in driving macrophage trained immunity [32]. Glycolysis, provides fast cellular ATP and fuels the TCA cycle whose metabolites and intermediates provides precursors for lipid biosynthesis essential for membrane remodelling and the synthesis of inflammatory mediators in M1 macrophages. TCA cycle metabolites such as fumarate, [33] and intermediates like succinate [34] regulate epigenetic reprogramming and macrophage proinflammatory phenotypes, while the TCA cis-aconitate metabolite when diverted to itaconate, exerts anti-inflammatory activity by inhibiting the succinate dehydrogenase-mediated oxidation of succinate to fumarate [35] (Figure 1).
In addition to mediators derived through glycolytic pathways, alterations in cholesterol metabolism can also play an important role in reprogramming and polarization of monocytes/macrophage [36]. Alterations in cholesterol metabolic pathways have been shown to regulate trained immunity not only in peripheral blood monocytes and tissue macrophages but also in bone marrow progenitor cells, indicating sustained transcriptional effects early in myeloid cell ontogeny [37]. Fatty acid and cholesterol synthesis is regulated at the transcriptional level by sterol regulatory element binding proteins (SREBPs) with the SREBP1-a isoform abundantly expressed in macrophages [38]. It is noteworthy that mice with Srebp1-a deficiency have defective innate immune responses with their isolated macrophages unable to induce lipid biosynthesis and a defective proinflammatory response to LPS [39]. Interestingly trained immunity in monocytes has recently been shown to be reversed using the cholesterol lowering agents statins [36].
While trained immunity contributes to mechanisms of immune protective responses during pathogenic insults it is increasingly recognized these same mechanisms may contribute to diseases with a chronic inflammatory component such as atherosclerosis. Support for this hypothesis is provided by studies describing the short-term treatment of apolipoprotein E-deficient (Apoe−/−) mice, with low doses of LPS to mimic chronic inflammation, which exacerbated the development of atherosclerosis while inducing sustained proinflammatory polarization of monocytes [40]. Further evidence of polarization of monocytes clinically is shown in patients with severe coronary artery disease (CAD), here a proinflammatory response to LPS is seen in circulating monocytes isolated from CAD patients compared to healthy subjects without atherosclerosis [41]. Other reports with isolated innate cells from patients again with atherosclerotic CAD link increased proinflammatory cytokine production with metabolic changes. Here increased glucose uptake and glycolytic flux raised IL-6 and IL-1β production from isolated innate cells in patients through activation of the enzyme pyruvate kinase M2 (PKM2) [42]. This occurs as PKM2 phosphorylates the transcription factor STAT3 which increases 1L-6 and IL-1 β at the transcriptional level [42].
Sophisticated imaging techniques such as intravascular ultrasound (IVUS) are capable of assessing features of high-risk vulnerable plaques in vessels [43] In the PROSPECT trial IVUS-identified thin caped fibrous lesion as an independent predictor of major adverse cardiovascular events in patients [44]. However, little is known about the metabolic state of plaque resident macrophages in humans, as studies are hampered by specific cell isolation techniques. To address this, Tomas et al. examined metabolomics in high-risk carotid artery plaques obtained by endarterectomy [45]. These plaques exhibited an increase at the transcriptional level of metabolic signature genes associated with glycolysis, amino acid utilization, and a decrease in fatty acid oxidation [45]. In addition, patients with acute coronary syndrome (ACS) show elevated levels of glycolytic metabolism using imaging analysis of fluorodeoxyglucose (FDG) uptake at atherosclerotic lesion sites compared to patients with stable angina [46]. Together, these findings indicate that metabolic changes leading to immune training of monocyte/macrophage cells may play a central role in driving atherosclerotic disease progression. The future use of such plaque imaging techniques, incorporating the macro to microscopic analysis, provide promise of being incorporated into the clinical setting to predict future CV events in patients [47].
Given the possible role of innate immune training in atherosclerotic disease, it is worth considering which specific factors present systemically and in the plaque microenvironment likely influence the metabolic rewiring of resident macrophages shaping their inflammatory function. As macrophage cells within the plaque die from metabolic and lipid stresses they release their contents which contributes to the necrotic core of the plaque. The plaque microenvironment overtime develops into a complex inflammatory lesion site characterised by elevated expression of proinflammatory cytokines, hypoxia, oxidised lipids, cholesterol crystals and danger associate molecular patterns (DAMPS) derived from dying cells [3]. Atherosclerosis severity is associated with systemic factors, such as hyperlipidemia, low-grade inflammation states associated with diabetes, autoimmune disease, and infection [15], all of which likely influence metabolic reprogramming in innate immune cells driving plaque progression [17] (Figure 1).
The necrotic environment of the plaque is an ample source of mediators that drive inflammation and potentially shape the metabolic responses of the innate cells involved. Oxidized phospholipids (OxPL) are a stimulus of innate immune training and present at high concentrations in atherosclerosis lesions. OxPLs mediate their effects by interaction with the scavenger receptor (CD36) on macrophage cells promoting macrophage foam cell formation [48] leading to activation of pro-apoptotic signalling in various cells, including smooth muscle cells (SMCs) [49] and macrophages [50]. It has been reported that endogenous OxPLs can potently boost the production of IL-1β, TNF-α and IL-6 by rewiring the metabolism of macrophages stimulated with LPS towards aerobic glycolysis [51]. Here the immune metabolic adaptations are also shown to occur in vivo in atherosclerotic mice, and pharmacological interference of OxPL driven metabolic changes reduced atherosclerotic plaque formation [51]. More importantly the transcriptional changes associated with the metabolic state induced by OxPL also occur in hypercholesterolemic individuals [51]. Diet influences in elevating OxPLs such as, administration of a western-type diet to Ldlr−/− mice also result in innate trained immunity through effects in bone marrow myeloid progenitor cells which persist following reversion to chow diet [52]. Interestingly, the western diet induced transcriptomic and epigenomic reprogramming of myeloid progenitor cells leading to enhanced innate immune responses was mediated via the NOD-like receptor pyrin domain–containing protein 3(Nlrp3) inflammasome. Additional studies in human monocytes also indicated that OxPL induced innate training is mediated by NLRP3 [52]. The NLRP3 inflammasome which regulates the activation of the proinflammatory cytokine IL-1β have been linked to various inflammatory diseases [53], including atherosclerosis [54] (Figure 1).
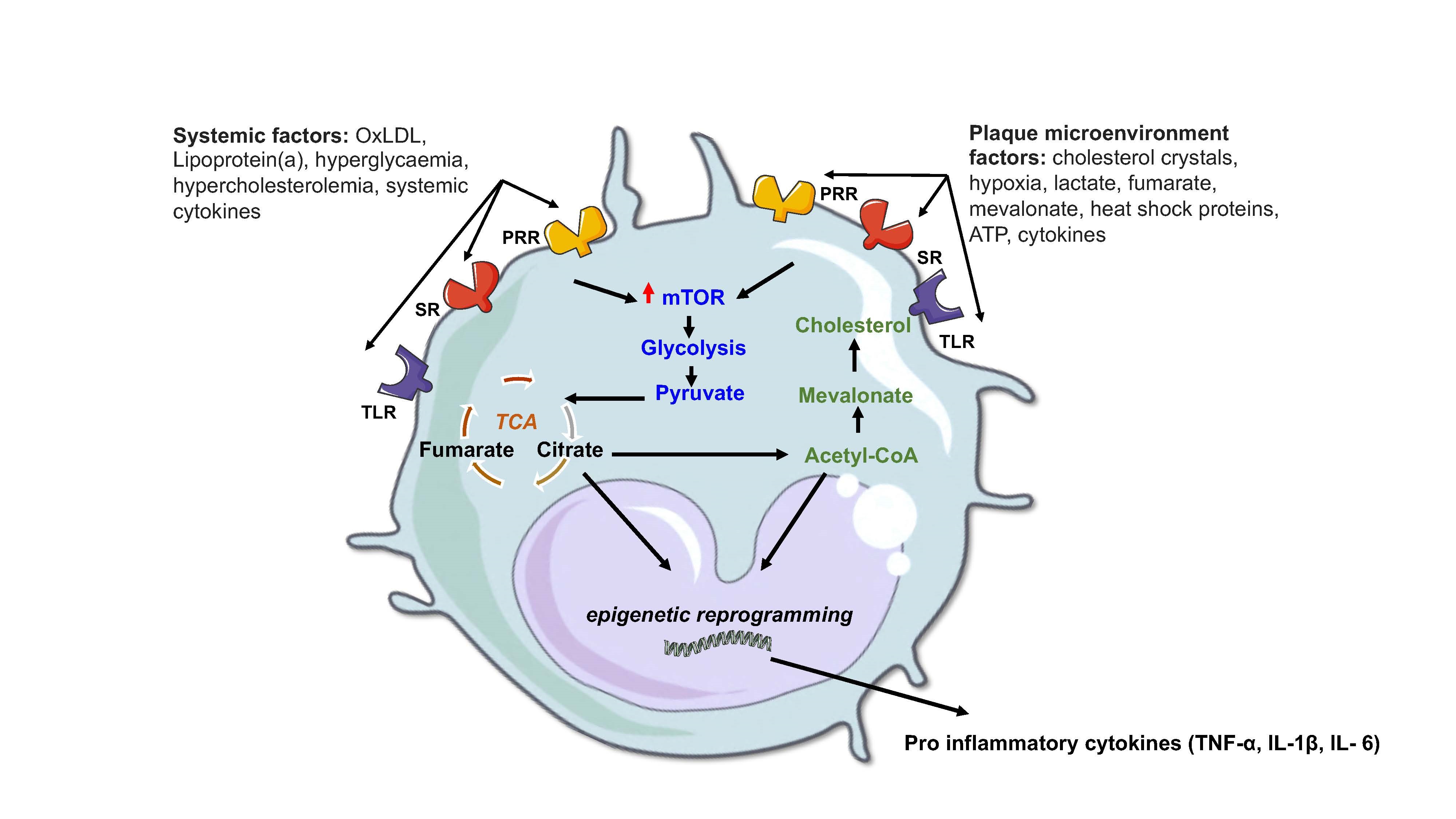 Figure 1. Atherosclerotic factors that modulate innate immune metabolic pathways. Pro atherogenic stimuli, either systemic or localized to the plaque microenvironment, engage with specific receptors such as pattern recognition receptors (PRR), toll-like receptors (TLR) and scavenger receptors (SR) in innate immune cells to induce a state of trained immunity associated with driving disease progression. This trained immunity involves an interplay between an increase in metabolic pathways such as glycolysis, a fragmented TCA cycle and cholesterol synthesis which lead to epigenetic reprogramming and enhanced gene transcription of disease associated cytokines as described in the text. Figure created using Servier Medical Art under a CC-BY Creative Commons license, smart.servier.com.
Figure 1. Atherosclerotic factors that modulate innate immune metabolic pathways. Pro atherogenic stimuli, either systemic or localized to the plaque microenvironment, engage with specific receptors such as pattern recognition receptors (PRR), toll-like receptors (TLR) and scavenger receptors (SR) in innate immune cells to induce a state of trained immunity associated with driving disease progression. This trained immunity involves an interplay between an increase in metabolic pathways such as glycolysis, a fragmented TCA cycle and cholesterol synthesis which lead to epigenetic reprogramming and enhanced gene transcription of disease associated cytokines as described in the text. Figure created using Servier Medical Art under a CC-BY Creative Commons license, smart.servier.com.
Hypercholesterolemic states result in disruption to cellular cholesterol homeostasis and can promote disease progression via the conversion of macrophages into proatherogenic hyperinflammatory cells [1]. The changes in lipid metabolism in macrophages is balanced by an influx and efflux of cholesterol critical to maintaining intracellular cholesterol homeostasis. Uptake of cholesterol by cells is primarily mediated trough the LDL receptor and the scavenger receptor CD36 [55], whereas the lipid transporters such as ATP-binding cassette transporter A1 (ABCA1) and G1 (ABCG1) are essential for cholesterol efflux [56]. The transcription factor sterol regulatory element binding protein 2 (SREBP-2) increases expression of genes involved in cholesterol uptake and biosynthesis when cellular cholesterol levels are low [57]. Whereas the transcriptional factor liver X receptor (LXR) increases expression of genes involved in cellular cholesterol efflux when cellular cholesterol is in excess [58]. Other cellular protective effects from excess free cholesterol within cells is conversion to cholesterol esters (CEs), with these CEs forming lipid droplets in macrophages giving rise to the familiar lipid laden foam cell at the plaque site.
While suppression of plasma cholesterol levels in patients with atherosclerosis has largely proven successful a residual risk of cardiovascular disease remains. Therefore unravelling and targeting the macrophage inflammatory component to this complex disease is of great interest. Hypercholesterolemia-induced disturbances of intracellular cholesterol metabolism may contribute a large component of this dysfunctional macrophage phenotype. Monocytes from patients with familial hypercholesterolaemia are characterized by enhanced cytokine production and epigenetic changes on the promoter regions of atherogenic cytokines, which can be detected even after 3 months of cholesterol-lowering statin treatment [59]. Macrophages are known to internalize OxLDL and other OxPLs leading to the formation of foam cells which accumulate in atherosclerotic plaques as described above [48]. This foam cell accumulation contributes over time to a chronic narrowing of the vessel lumen, as well as vessel wall endothelial dysfunction and vascular smooth muscle migration. Macrophages exposed to OxLDL have an enhanced response to restimulation with TLR ligands to produce IL-6 and TNF-alpha with an increased propensity to form foam cells [60,61]. During OxLDL exposure these macrophages increase lipid scavenger receptor expression (CD36 and SR-A) facilitating further OxLDL uptake with a reduction in cholesterol efflux receptors (ABCA1 and ABCG1). Mice with defective cholesterol efflux transporters ABCA1 and ABCG1 in the myeloid lineage are characterized by a greater expansion and proliferation of hematopoietic stem cells (HSCs) [62]. In addition, during the induction of trained immunity upon exposure to OxLDL, there is an activation of the cholesterol synthesis pathway in macrophages [27]. While studies had previously revealed that intracellular cholesterol loading increases the glucose transporter GLUT1 expression, activating glycolysis, and oxidative phosphorylation to produce a hypermetabolic state in leukocyte and myeloid cells [63].
In this regard it is perhaps unsurprising that targeting lipid metabolism in macrophages has been found to improve atherosclerosis outcomes in mice models. The overexpression of LXRα, in macrophages resulted in an antiatherogenic effect in a murine models of disease by increasing cholesterol efflux in a macrophage cell specific fashion [64]. Similarly, it has been demonstrated that treatment with an LXR agonist results in attenuation of atherosclerosis in several murine disease models [65]. Importantly, individuals with upregulated macrophage LXR expression have also been reported to be less susceptible to the development of atherosclerosis [66]. Together these findings reinforce the hypotheses linking hyperlipidemia and cellular cholesterol levels as key pathogenic drivers of chronic vascular disease progression.
Altered cellular metabolism in macrophages from stimuli residing in the plaque microenvironment as well as systemic mediators likely contributes to their cellular function across all stages of atherosclerosis, from lesion initiation, to formation of advanced lesions characterized by necrotic cores, to lesion regression following aggressive lipid lowering (Figure 1).
The clinical benefit of statin agents have long been described not only for their ability to lower plasma cholesterol, but also for modulation of inflammatory processes. This anti-inflammatory effect was first inferred from several statin clinical trials where a greater level of cardiovascular benefit was observed which did not entirely correlate with the degree of the drugs cholesterol lowering ability [67]. These observations, alongside the emerging understanding that atherosclerosis is recognized as an inflammatory disease [68] coincided with studies detailing the effects of statins on reducing inflammation. Early studies in mouse models of atherosclerosis, high doses of statin therapy were observed to attenuate the accumulation of aortic cholesterol without altering plasma lipid levels [69]. While the need for systemic high doses of statin agents to impact inflammation at the plaque site can result in a greater risk of unwanted side effects, this has been overcome by targeted macrophage delivery of statins using nanoparticles which accumulate in the plaque microenvironment [70]. Both a low and high dose statin nanoparticle treatment regimen in mice inhibited plaque inflammation progression [70].
The observation that statins can exhibit immunomodulatory functions have also prompted broader investigations into their potential therapeutic properties [67]. Studies have shown that statins can improve survival from sepsis in mice [71] and a meta-analysis summarizing the effects of statins on mortality in patients with infection and/or sepsis seems to support the hypothesis of a protective effect [72]. However, definitive conclusions here cannot be drawn due to considerable heterogeneity across the studies including types of statins, dosages, and duration of statin administration. Observational studies in other inflammatory autoimmune disease states such as in multiple sclerosis [73] and rheumatoid arthritis [74], have also indicated disease activity and inflammation is reduced in patients receiving statin treatment. In support of the potential anti-inflammatory clinical benefit above mechanistically, statins have also been shown to reduce the activity of transcription factors such as nuclear factor kB (NF kB ) and activator protein 1 (AP-1), which are implicated in the regulation of a wide range of atherosclerotic inflammatory pathways [75]. Statins also exert direct anti-inflammatory effects on a range of cells that are involved in atherosclerotic plaque development and rupture [76]. Importantly, pathways that play a key role in the inflammation driving atherosclerosis are downregulated by statins such as NLRP3 inflammasome, cathepsin B, and their downstream mediators IL-1β and IL-18 [77]. More recently, it has also been demonstrated that administration of statins to patients inhibits the subsequent expression and activation levels of IL-1β induced by CC in innate immune cells [9]. In contrast others have reported statins can exhibit a stimulatory effects on the NLRP3 inflammasome in vitro, leading to the secretion of IL-1β and IL-18 [78] while a study with high-dose atorvastatin demonstrated no anti-inflammatory effects in normolipidemic subjects [79]. These contradictory inflammatory profiles, observed with various statins to date, may be related to differences in their chemistry and pharmacokinetics properties, as well as baseline plasma lipid levels in various patient groups studied. Hydrophilic statins such as pravastatin and fluvastatin do not easily penetrate the cellular membrane resulting in less pleiotropic properties, unlike lipophilic statins such as simvastatin, lovastatin, and atorvastatin which exhibit greater cellular uptake and more pleiotropic effects reported [80]. Whether these immunomodulatory effects contribute significantly to protection from atherosclerosis, independently of lipid lowering, remains to be established.
Membranes of cells and organelles are rich in cholesterol where it is important for maintaining structural integrity and fluidity of the membrane as well as signal transduction mechanisms. As such, cholesterol plays many important roles in facilitating immunological responses from endocytosis of foreign bodies, to cellular growth and proliferation as well as many other cellular processes [81]. As a primary mechanism of action, statins inhibit the rate-limiting enzyme of the cholesterol synthesis pathway, thereby reducing mevalonate as well as downstream isoprenoid intermediates that are required for prenylation reactions [82]. Isoprenoid intermediates of the cholesterol synthesis pathway are used for the prenylation of signalling molecules, accommodating their integration into the cell membrane and allowing transduction of important immunological processes. Modulation of these cellular effects, downstream of cholesterol synthesis, have been proposed as a mechanism of action through which statins can modulate various immune responses including cellular signalling, antigen presentation, immune cell migration and cytokine production [83].
Recent observations identifying the central role of the cholesterol synthesis pathway in the induction, activation and/or amplification of innate trained immunity are also of significant relevance to statins immune effects [52]. Interestingly, cholesterol inhibition by statins was found to inhibit increased cytokine production and epigenetic reprogramming induced by both β-glucan and oxLDL [36]. Central to this effect was the reduction in accumulation of the metabolite mevalonate rather than the levels of cholesterol or reduction in prenylation reactions [36]. Mevalonate was subsequently shown to act as a mediator of innate immune training via increasing expression of the insulin-like growth factor 1 (IGF1)- receptor and activation of mTOR signalling pathways, leading to subsequent histone modifications and epigenetic changes in inflammatory pathways [36].These data point towards an important role for the intracellular cholesterol/mevalonate synthesis pathway in the induction of innate trained immunity. Studies in murine models above were further validated in patients with a hereditary autoinflammatory syndrome (HIDS). HIDS patients carry mutations in mevalonate kinase which leads to an accumulation of mevalonate and is likely responsible for the clinical presentation of disease characterised by exaggerated IL-1β production by monocytes leading to febrile episodes [84]. Inhibition of IL-1 receptor signalling with anakinra (IL-1 receptor antagonist) is a successful therapy for these HIDS patients [85]. A trained immunity phenotype, in monocytes from HIDS patients as demonstrated by exaggerated cytokine production, transcriptional and epigenetic changes is accompanied by mevalonate accumulation [36]. Interestingly, a previous study with statin treatment reduced mevalonate levels in HIDS patients, and decreased the number of febrile days [86], supporting the concept that lowering mevalonate levels is beneficial for disease activity in this setting. These data provide a novel link between the immunosuppressive effects of statins and their impact on immune cell metabolism which may have important implications for their atherosclerotic protective function (Figure 2).
While statins targeting the cholesterol synthesis pathway have proven to be effective in preventing trained immunity in vitro, it is noteworthy that they failed to revert the proinflammatory state of monocytes from patients with familial hypercholesterolemia (FH) [59]. The proinflammatory state seen with monocytes from patients with FH can be described as a trained immune phenotype with proinflammatory epigenetic modifications. It is possible that in such patients a prolonged state of hypercholesterolemia induces myeloid progenitor reprogramming that can persist and is insensitive to statins effects, as previously described for Ldlr−/− mice exposed to a western high fat diet [52]. It will be interesting to see if agents such as bempedoic acid, which also target the cholesterol synthesis pathway and limits formation of the metabolite mevalonate, has similar effects on trained immunity as demonstrated with stains. It will be equally interesting to investigate the potential for immunomodulatory effects with other novel cholesterol lowering therapeutic approaches particularly aimed at familial hypercholesterolaemia such as PCSK9i as well as approaches aimed at inhibiting synthesis of lipoproteins such as lomitapide and mipomersen. These latter two agents, while effective at lowering LDL-C, do so without directly inhibiting cholesterol synthesis and therefore may be less likely to impact the metabolite mevalonate. There remains a continued need in utilising the lipid lowering therapies to explore their full potential as targets for the proinflammatory and immunometabolic component of atherosclerosis pathogenesis. Such efforts are underscored by the establishment of proof of principle that inflammation and trained immunity is an important driver of disease from recent clinical studies [4,87].
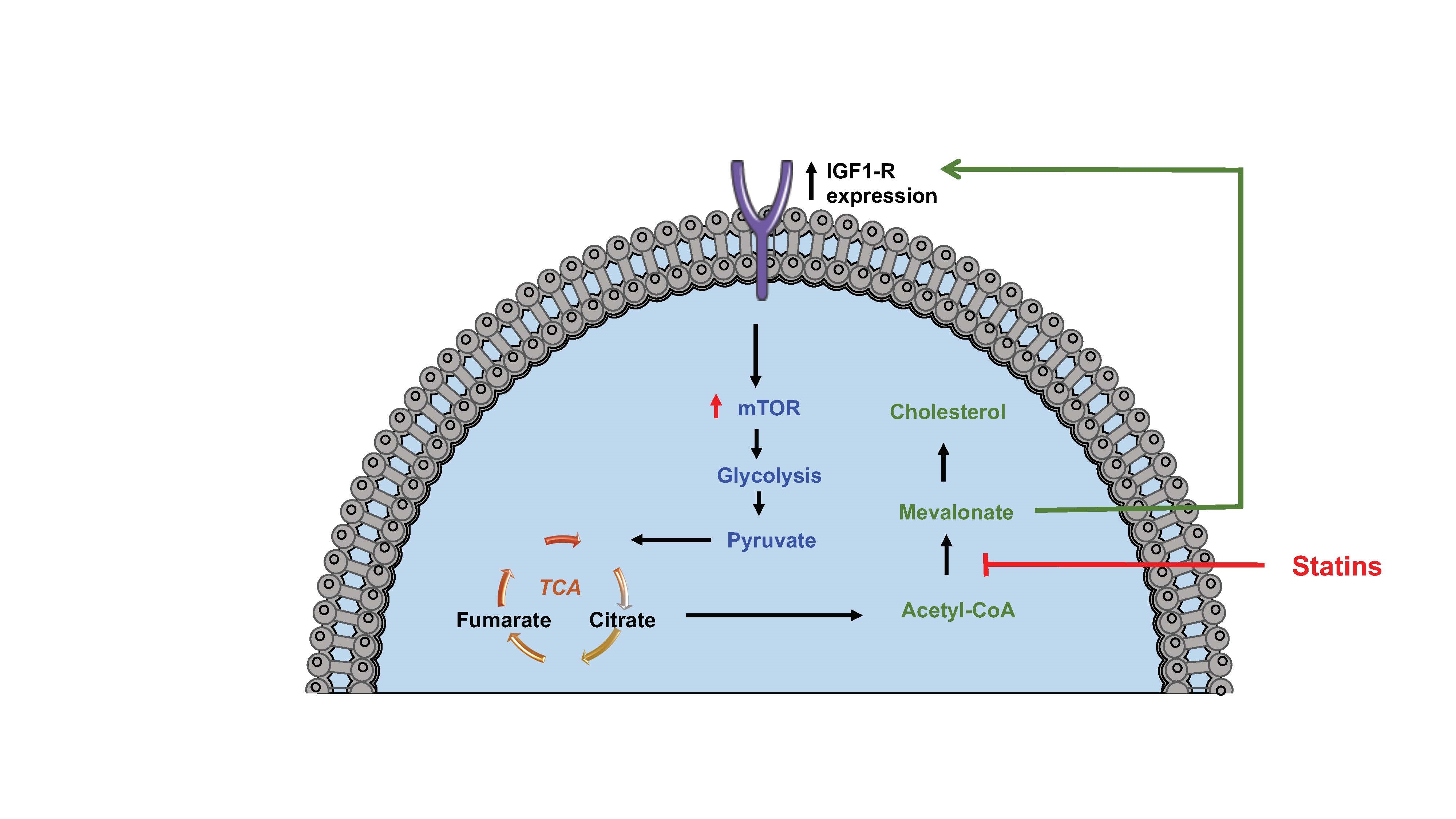 Figure 2. Statins inhibit glycolysis in monocyte trained immunity. Blocking cholesterol synthesis using statins inhibits the production of mevalonate which is a driver of IGF1-R expression induced aerobic glycolysis and trained immunity. Figure created using Servier Medical Art under a CC-BY Creative Commons license, smart.servier.com.
Figure 2. Statins inhibit glycolysis in monocyte trained immunity. Blocking cholesterol synthesis using statins inhibits the production of mevalonate which is a driver of IGF1-R expression induced aerobic glycolysis and trained immunity. Figure created using Servier Medical Art under a CC-BY Creative Commons license, smart.servier.com.
Clinically, the discovery of a role for mevalonate in the induction of trained immunity, has important consequences for the chronic inflammatory state associated with atherosclerosis. In particular inhibition of innate cellular mevalonate through manipulation in the cholesterol synthesis pathway by potent localised delivery of statins to lesion sites or with novel inhibitors of mevalonate signalling may offer an effective approach to targeted therapy of the hyper-inflammatory component in which this trained immunity plays a role [88].
Macrophage cells play a crucial role in the immune responses associated with atherosclerosis. Growing evidence indicates that metabolic pathways contribute to the trained immunity and epigenetic programming of these innate cells for their activities. While oxidative metabolism seems to be required for suppressive immune macrophage function, glycolysis and lipid metabolism is closely associated with their proliferation and migration. Crucial drivers of atherosclerosis such as hypercholesterolemia may disturb macrophage metabolism and drive disease progression. Statins, as small molecule inhibitors of cellular cholesterol synthesis, can alter these proinflammatory metabolic signatures to potentially lessen these disease modifying effects during atherogenesis. Additional pathways through which cholesterol synthesis may influence immunity cannot be overlooked such as changes in membrane fluidity which may also effect signalling transmission. The different approaches to date have demonstrated that statins affect the immune response at multiple levels including signalling, gene transcription, and now epigenetic modifications via immune metabolism and trained immunity. In addition, the immunomodulatory effects of statins in different disease models are underscored by robust data from multiple independent groups, as well as indicating that modulation of cellular metabolic pathways holds promise for targeting the innate immune training aspect of inflammation. It is also worth noting that statins exhibit a relatively low immunosuppressive potency when compared to many established immunosuppressive drugs. However, the immune suppressive effects with statin use may prove advantageous when approaches to immune modulation rather than strong immunosuppression is investigated and developed particularly in settings such as atherosclerosis. Future research aimed at identifying novel metabolic pathways that direct macrophage immune function in atherosclerosis, as well as strategies to inhibit such pathways may offer future therapeutic approaches.
The authors declare that they have no conflicts of interest.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
80.
81.
82.
83.
84.
85.
86.
87.
88.
Lioudakis E, Lucitt M. Statin Disruption of Cholesterol Metabolism and Altered Innate Inflammatory Responses in Atherosclerosis. Immunometabolism. 2021;3(3):e210023. https://doi.org/10.20900/immunometab20210023

Copyright © 2020 Hapres Co., Ltd. Privacy Policy | Terms and Conditions




