Location: Home >> Detail
J Psychiatry Brain Sci. 2023;8:e230012. https://doi.org/10.20900/jpbs.20230012
 ,
Xiaogang Chen 1,2,*,
,
Xiaogang Chen 1,2,*,
1 Department of Psychiatry, National Clinical Research Center for Mental Disorders, and National Center for Mental Disorders, The Second Xiangya Hospital of Central South University, Changsha Hunan 410011, China
2 China National Technology Institute on Mental Disorders & Hunan Key Laboratory of Psychiatry and Mental Health, Mental Health Institute of the Second Xiangya Hospital, Central South University, Changsha, Hunan 410011, China
* Correspondence: Zongchang Li; Xiaogang Chen.
Bipolar disorder (BD) is one of the major psychiatric disorders, which damages the cognition, emotion, and physical activity of patients and causes a huge financial burden on families and society. As an important epigenetic mechanism, DNA methylation is stable and reversible and has been considered to be implicated in the development of bipolar disorder through regulating gene expression. Currently, the detection of abnormal DNA methylation positions or regions in BD is mainly based on candidate genes and epigenomic-wide DNA methylation profiling. For example, many researchers have focused on abnormal DNA methylation of BD risk genes, such as COMT, SLC6A4, BDNF, and CACNA1C. In epigenomic-wide DNA methylation association studies, researchers have identified a few novel BD-associated aberrant DNA methylation positions or regions by improving experimental design, including more subjects, and using new DNA methylation detection technologies. In addition, DNA methylation editing technologies can be used to precisely manipulate DNA methylation and de-methylation in vivo or in vitro and uncover how DNA methylation affects gene transcription, which can be used to scientifically clarify the epigenetic mechanisms underlying the pathogenesis of BD in future studies.
Bipolar disorder (BD) is a severe neuropsychiatric disorder with lifetime and 12-month prevalence rates of 2.4% and 1.5%, respectively [1]. There are two main types of BD: bipolar I disorder (BD I) and bipolar II disorder (BD II). BD I is defined by the presence of typical manic episodes during which symptoms, such as overconfidence, grandiosity, talkativeness, insomnia, and even delusions and hallucinations, occur [1]. BD I patients may experience hypomanic episodes, depression episodes, and mixed episodes [2,3]. BD II is characterized by milder mood elevation during hypomanic episodes and severe mood distress during depression episodes [1]. BD II patients do not have manic episodes, but mixed episodes can occur [2,3]. Compared to patients with mania, those with hypomania have relatively intact judgement and shorter and milder symptoms [2]. Patients with depression often experience sadness, decreased energy, social withdrawal, hypersomnia, low self-esteem, and even suicide [2]. In addition, mixed episodes are acute episodes of mood alterations that can include both depressive symptoms and manic symptoms [2]. BD tends to occur in the early 20s, and an earlier onset is not a good thing [1,4]. Without regular treatment, the patient's mood episodes will be pronounced as the disease progresses, from threshold syndrome, then to recurrent depressive and (hypo)manic episodes, and finally to persistent and uninterrupted illness [4]. These always result in impaired interpersonal relationships, disabled social function, high rates of co-morbidity, and increased mortality for the patient [1]. According to the Global Burden of Disease Study 2019 (GBD 2019), BD accounted for 8.52 million disability-adjusted life years (DALYs), accounting for 0.33% of total DALYs and ranking 67th out of all 369 diseases and injuries [5]. Therefore, it brings enormous economic loads on families and society, and accurate diagnosis and effective treatment of patients is an area where research is urgently needed.
In recent years, in the field of epigenome research, the pathogenic mechanisms of an increasing number of diseases have been uncovered, which facilitates the development of new diagnostic and therapeutic approaches. Environment, especially the early life environment, can determine how genes are turned on and off through epigenomic modification. BD was considered a distinct and highly heritable disorder by family and twin studies [6], and several environmental events (such as infections during pregnancy, childhood trauma, and chronic stress) have been reported to be associated with higher morbidity of BD [7–10]. Therefore, it is well established that genetic and environmental factors play an interactive role in the development of BD by mediating epigenetic regulation [7–10]. DNA methylation is one of the most widely studied and important epigenetic modifications, which can regulate gene expression beyond changing DNA sequence, and play an important role in some basic biological processes, such as cell proliferation and differentiation and early embryogenesis [11]. Because of its heritability and reversibility, in the field of psychopathology, DNA methylation studies are gradually emerging. In this review, we summarized the main findings of DNA methylation studies for BD, including global DNA methylation, genome-wide DNA methylation, and site-specific DNA methylation. Then, we discussed that methylation-related multi-omics data integration analysis can be used in the studies of the functional mechanisms of DNA methylation in BD. Finally, we discussed the possibility of using cellular and animal experiments to study DNA methylation positions (DMPs) or DNA methylation regions (DMRs) based on previous studies. For example, we discussed the possibility of DNA methylation editing of candidate DMPs or DMRs to precisely study the pathogenesis of BD.
The systematic literature search of PubMed and Embase (Elsevier) datasets was performed using the keywords “bipolar disorder”, “bipolar affect disorder”, and “DNA methylation” limiting the date from February 2013 to January 2023. Considering the urgency of the topic and to increase the sensitivity of the search, a gray literature search was conducted using Google Scholar.
Inclusion and Exclusion CriteriaWe only chose articles published in English and peer-reviewed publications and excluded graduate theses and dissertations. There were many types of studies discussed in this review, including randomized controlled trials, cohorts, and cross-sectional studies.
DNA methylation is one of the most widely studied and predominant epigenetic modifications. Compared with other mechanisms (such as histone modification), it is more stable and can be inherited from parents to offspring [12]. DNA methylation usually occurs on the cytosine of the CpG dinucleotides. In recent years, some sensitive technologies have been used to interrogate DNA methylation patterns in different types of cells. Moreover, in the field of bioinformatics, considerable amounts of multi-omics data can be exploited. Integrating analysis of methylomics and other omics data (such as genomics and transcriptomics data) can greatly improve our knowledge of the functional mechanisms of DNA methylation. In addition, epigenome editing technologies have developed to precisely regulate DNA methylation in CpG domains, suggesting the possibility that researchers further investigate the role of specific DMPs.
By regulating gene expression and repressing transposons, DNA methylation plays diverse roles in mammalian development and disease [13]. DNA methylation profiling and epigenome editing technologies can be used to discover and verify differences in DNA methylation between patients and healthy controls and thus explore the molecular biological mechanisms of disease development. Therefore, a large number of studies have been performed to explore the potential roles of DNA methylation in BD by using various designs or methods, including global DNA methylation, genome-wide DNA methylation, and site-specific/regional DNA methylation profiling, DNA methylation-related multi-omics data integration analysis and DNA methylation-related experiments.
DNA methylation and demethylation require the involvement of different enzymes. The normal cytosine nucleotide in DNA is converted to 5-methylcytosine(5mC) by DNA methyltransferase (DNMT), by transferring the methyl group from S-adenosyl-methionine onto the fifth carbon of cytosine [14]. There are three main types of DNA methyltransferases: DNMT1, DNMT3A, and DNMT3B [14–16]. DNA demethylation requires enzymatic reactions to convert 5mC to naked cytosine. A well-known DNA demethylation mechanism is mediated by the ten–eleven translocation (Tet) enzymes, including Tet1, Tet2, and Tet3 [17,18]. Tet can add hydroxyl groups onto 5mC to form 5hmC that is then further converted to naked cytosine. When located in a gene promoter, DNA methylation is typically associated with gene silencing [19–21]. In contrast, DNA methylation in the gene body may activate gene transcription [22]. Therefore, the inactivation of each of these genes can have disastrous consequences [16,23].
BD has been shown to be associated with the dysregulation of DNMTs, particularly DNMT1. DNMT1 expression has been found to be significantly reduced during depressive episodes, but not during the remissive states of BD [24], and negatively correlated with the expression of glutamic acid decarboxylase67 (GAD67), reelin (RELN), and brain-derived neurotrophic factor (BDNF) in the brain of BD [25]. A study has detected that deletion of DNMT1 in forebrain neurons in mice may cause anxiolytic- and antidepressant-like effects [26], suggesting that modulation of DNMT1 may produce therapeutic effects in BD.
DNA methylation age is termed the “epigenetic clock” and is correlated with chronological age [27,28]. Among the six potential biological age predictors (epigenetic clocks, telomere length, predictors based on transcriptomic, proteomic, metabolomics, and composite biomarkers), epigenetic clocks have been identified as the most promising biomarkers [29]. Moreover, DNA methylation age can predict age-related conditions and lifespan [28], and is also associated with various psychiatry diseases, such as schizophrenia (SCZ) [30–33], major depression disorder (MDD) [34], BD [35] and autistic disorder [36]. DNA methylation age studies on BD were summarized in Table 1.
Because of its high mortality rate resulting from age-related medical conditions (such as cardiovascular disease [37]), BD is considered a disease of accelerated aging [38,39]. Converging evidence has indicated accelerated epigenetic aging in patients with BD. A study has found that older patients with BD presented significantly higher epigenetic aging acceleration (EAA) in the blood than healthy controls, but did not in younger BD patients [35]. However, this finding was not replicated in the cerebellum of patients with BD [35]. Another study has divided the sample by median age and found higher EAA in the hippocampus of older patients with BD compared to age-matched healthy controls [40]. Furthermore, a study has found that BD had a higher EAA regardless of individual age [41]. In addition, Okazaki S. et al. found that EAA was significantly decreased in patients with BD with long-term and high usage rates of mood stabilizers, including Li, sodium valproate, and carbamazepine [42]. In addition, environmental factors (such as socioeconomic position, childhood trauma, lifestyles, and psychosocial stress) have been considered to have a significant effect on the etiology and clinical course of BD [43], which are also linked to epigenetic aging acceleration [44].
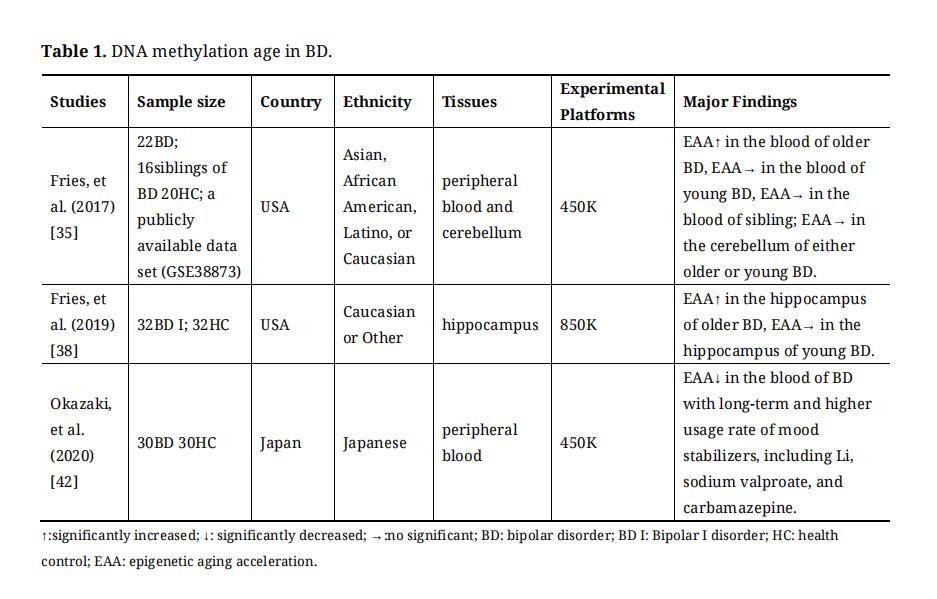 Table 1. DNA methylation age in BD.
Table 1. DNA methylation age in BD.
In summary, DNA methylation age may be a proxy for understanding the pathophysiology of BD, such as the onset and severity of disease, remission and relapse, and response to medication. In addition, considering the above results, the effect of environmental factors, medical treatment, and age-related medical conditions on BD may be explained by EAA, which should be considered in future large sample and longitudinal studies.
Measuring global DNA methylation (GMe) was only able to compare general methylation levels between the case and control groups. Many studies are focusing on GMe in psychiatry disorders [45–53]. GMe can be interrogated by several technologies. For example, high-performance liquid chromatography-ultraviolet (HPLC-UC) and pyrosequencing of Long interspersed nuclear elements-1 (LINE-1). HPLC-UC is the“gold standard” method to calculate the ratio of methylcytosines to genome-wide cytosines [54]. However, the disadvantages of HPLC-UC limit its use, including the need for specialized laboratory equipment and relatively large amounts of DNA samples (3–10 µg) [54]. LINE-1 is a genomic repetitive element accounting for 17% of the whole genome [55]. Pyrosequencing of LINE-1 can be used as the easiest and most convenient proxy for HPLC to estimate GMe [55]. In addition, enzyme-linked immunosorbent assay (ELISA), luminometric methylation assay, pyrosequencing of Alu repeat elements, and restriction enzymes-based method can also be used for measuring GMe [54,55]. Global DNA methylation studies on BD were summarized in Table 2.
The results of GMe studies in BD were discrepant. The GMe levels in BD have been shown to be below or unchanged compared to controls, and be affected by medication. Through the examination of blood cell samples, some studies revealed that GMe was unchanged in bipolar patients compared to healthy controls [50,56], and GMe in BD wasn’t associated with mood episodes, illness duration, number of manic episodes, number of depression episodes, and lifetime psychotic symptoms [57]. Pharmacological studies of BD suggested that different pharmacological treatment strategies were associated with different global methylation levels in patients with BD. A study has indicated that transformed lymphoblasts of BD with an excellent response to lithium monotherapy have lower GMe levels, and lithium treatment will further reduce GMe of BD [52]. Second-generation antipsychotic (SGA) users have displayed a lower global methylation level compared with mood stabilizer users, but valproic acid (VPA) treatment hasn’t significantly influenced GMe in the SGA or mood stabilizer group [58]. Another study has also found that BD on Li monotherapy had significantly decreased GMe compared with both controls and BD on lithium + valproate treatment [59]. According to these studies, global methylation level in BD was significantly influenced by medicine treatment [52,56,58,59], mood states [50,56], insulin resistance [58], smoking [50], and different body tissues [52,59]. There are a lot of unknowns within the relationship between GMe with these factors, which need to be explored. Furthermore, GMe cannot reflect the quantification of differences in genome-wide or specific-site DNA methylation differences that may provide more information. Therefore, more and more researchers have focused on genome-wide DNA methylation and Candidate gene DNA methylation in BD.
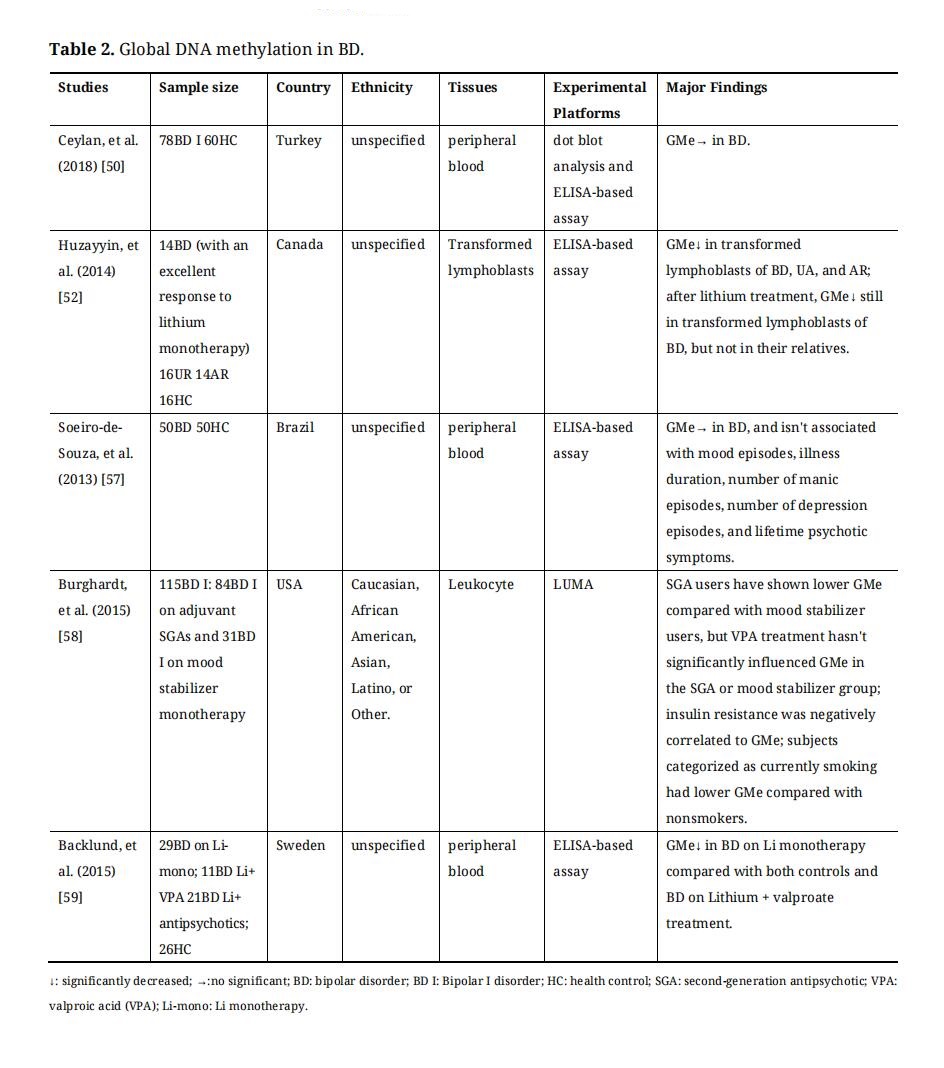 Table 2. Global DNA methylation in BD.
Table 2. Global DNA methylation in BD.
Main genome-wide DNA methylation profilings are based on microarray hybridization and next-generation sequencing platforms [60–62]. Before profiling DNA methylation, several methods are available for the initial processing of DNA samples, including electrophoresis-based methods, affinity enrichment-based methods, restriction enzymes-based methods, and bisulfite conversion-based methods [61]. Recently, the most widely used microarray hybridization technology is Illumina Infinium HumanMethylationEPIC BeadChip (850K), which is cost-effective [62]. Next-generation sequencing has emerged in recent years, which contains whole genome bisulfite sequencing (WGBS), reduced representation bisulfite sequencing (RRBS-Seq), methylated DNA immunoprecipitation deep sequencing (MeDIP-seq), genome-wide methylomic approach (SeqCapEpi), etc. [54,60,61,63]. With the development of single-cell technology, genome-wide DNA methylation can be studied at the single-cell level [64]. Genome-wide DNA methylation studies on BD were summarized in Table 3.
Along with the rapid development of technology, using genome-wide DNA methylation studies in BD, the researchers have found a few meaningful results. By using 850K, a study identified greater numbers of DMPs between the dorsolateral prefrontal lobe (DLPFC) and temporopolar cortex (TP) in BD, MDD, and healthy controls, and then detected many group-specific and group-overlapped DMPs or DMRs among the three groups [65]. The genes and pathways of methylomic differences were mainly involved in axon guidance, glutamatergic and GABAergic neurotransmissions, synaptic plasticity, and various neurodevelopmental mechanisms [65]. Another study in neuronal and nonneuronal nuclei in the PFC of BD has found promoter-wide DNA hypomethylation in BD [66]. Using gene ontology (GO) analysis, this study has also found that DMR-related genes were enriched in neurons and/or non-neurons for some biological processes, including hypomethylated molecular motor-related genes in neurons, chemokines in both cell types, and ion channel and transporter-related genes in non-neurons [66]. For the DNA methylome in human microglia, a study has identified various DMRs associated with mood disorder status [67]. These DMRs are highly related to microglial immune functions, circadian rhythms, or neuropsychiatric disorders [67]. In addition, using 450K, 850K profiling and PRS (polygenic risk scores), a DMP in VARS2 has been found to be most significantly hypomethylated in individuals with a high polygenic burden for BD [68]. Then, some BD-related genes, including MLC1, ESR1, KCKN5, L1CAM, CPEB1, and GABBR2, have mapped to nominally significant DMPs [68].
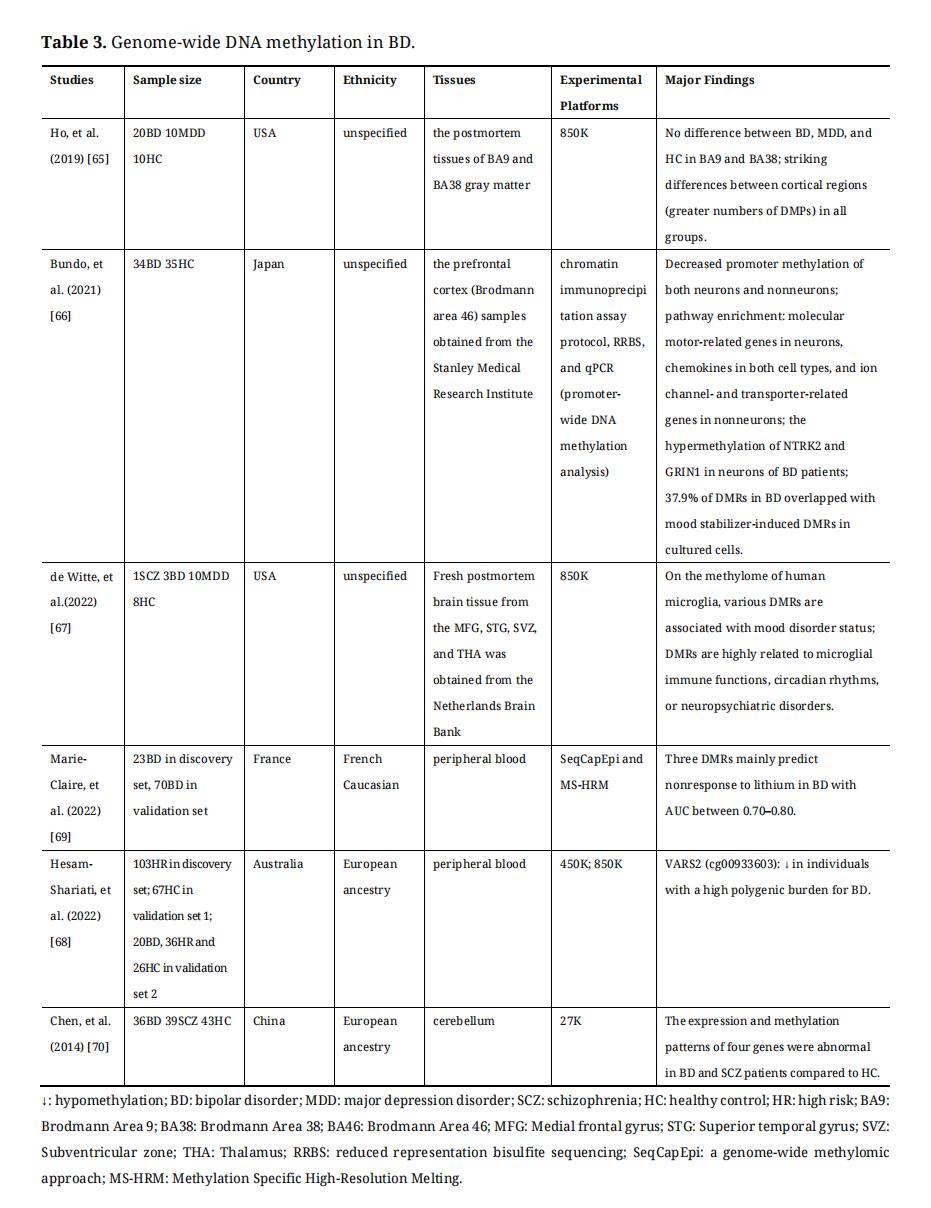 Table 3. Genome-wide DNA methylation in BD.
Table 3. Genome-wide DNA methylation in BD.
Furthermore, DMRs or DMPs have been identified using genome-wide DNA methylation profiling, which can be used to explore the role of DNA methylation in the diagnosis and treatment of BD in future studies. For example, mood stabilizers can change DNA methylation in the opposite direction and 37.9% of DMRs have been reported in BD patients overlapping with mood stabilizer-induced DMRs in cultured cells [66]. There was one of the most recent studies focused on methylomic biomarkers of lithium response in BD [69]. This study has found seven DMRs that discriminated responders from non-responders using SeqCapEpi in the first step and has verified three out of seven DMRs which mainly predicted nonresponse with AUC between 0.70–0.80 by using Methylation Specific High-Resolution Melting (MS-HRM) in the second step [69].
In addition, genome-wide DNA methylation data can be integrated with gene expression data for analysis. A study has combined genome-wide DNA methylation profiling with gene expression profiling and found 4 genes with differential association between expression and methylation patterns in the brains of BD and SCZ patients compared with healthy control [70].
In summary, genome-wide DNA methylation profiling in BD can identify novel BD-related DMRs or DMPs in different brain regions and cell types. These DMPs/DMRs are associated with specific genes, biological pathways, and biomarkers. Therefore, it may provide some basis for further exploration of the pathophysiology of BD. In future studies, genome-wide DNA methylation profiling studies of BD will need to be performed with larger sample sizes and on different treatment strategies. In addition, single-cell DNA methylation profiling may be useful in our understanding of the biological mechanisms of DNA methylation in BD.
Specific-loci DNA methylation associated with BD will be discussed in the following section. It can be measured by many methods, including bead array, polymerase chain reaction (PCR) and sequencing, pyrosequencing, methylation-specific PCR, and PCR with high-resolution melting (HRM-PCR) [54]. Specific-loci DNA methylation has been widely studied in different genetic regions associated with the biological systems underlying the pathophysiology of BD, such as central neurotransmitters, neuroendocrine, and immune systems. Candidate gene DNA methylation studies on BD were summarized in Table 4.
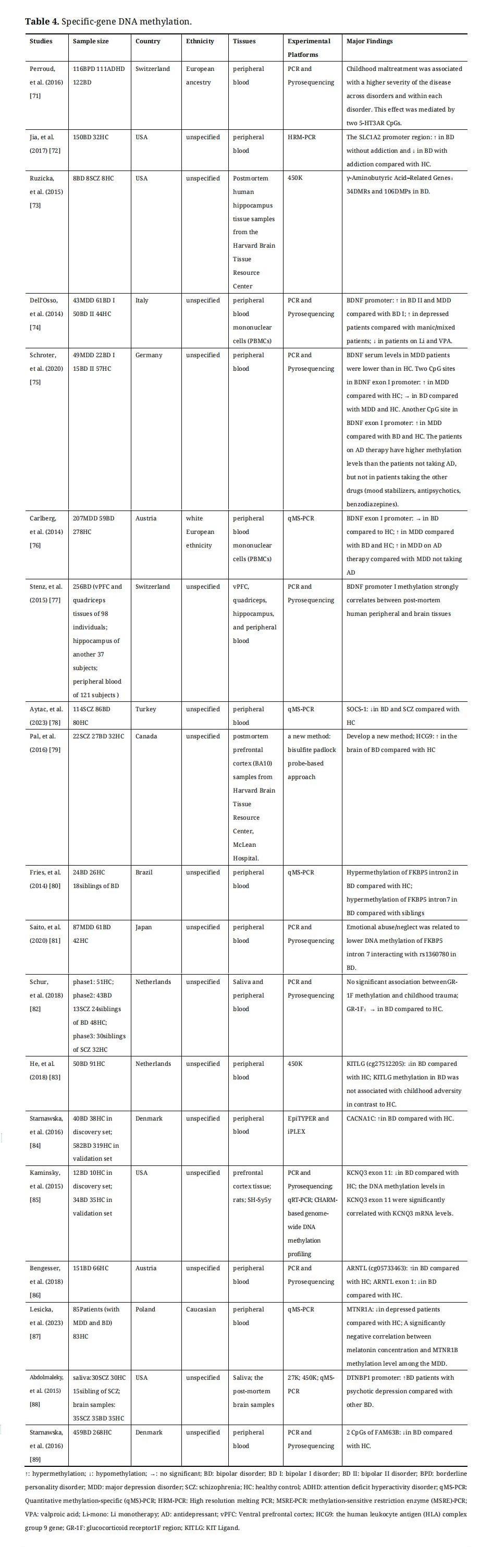 Table 4. Specific-gene DNA methylation.
Table 4. Specific-gene DNA methylation.
Abnormalities in central neurotransmitter function, which is implicated in the pathophysiology of BD, have received widespread academic attention. The main abnormal neurotransmitter systems in BD include the dopaminergic (DA) system, the serotonergic system [90,91], the glutamatergic system, and the gamma-aminobutyric acid (GABA)-ergic system.
The dopamine hypothesis of BD suggests increased dopamine levels during manic episodes and decreased dopamine levels during depression episodes [90,91]. The catechol-O-methyltransferase (COMT) is involved in the pathway of DA degradation, which has two forms: soluble (S-COMT) and membrane-bound (MB-COMT). DNA hypomethylation of MB-COMT was identified in both the brain and saliva of BD with high levels of gene expression [92,93], while overall COMT methylation levels were not altered in FC of BD [94]. In addition to COMT, DNA methylation levels of many other DA system genes (such as DA receptors (DRDs) and DA transporter (DAT)) need to be investigated in future studies of BD.
Serotonin, or 5-hydroxytryptamine (5-HT) has wide functions in the brain, which can regulate mood, cognition, reward, learning, and memory. The role of genetic variations in the serotonergic system in the pathogenesis of BD has been widely researched in previous studies [95–99]. The methylation of serotonin receptors (including HTR1A, HTR2A, and HTR3AR) and serotonin transporter (SLC6A4) genes has been studied in BD [100]. Hypermethylation of the HTR1A promoter has been identified in blood leukocytes of BD and SCZ by using HRM [101]. Genotype-dependent methylation levels in the promoter and introns of HTR2A were abnormal in the brain and saliva of BD [102,103]. Two 5-HT3AR CpGs have been shown to mediate the positive correlation between childhood maltreatment and the severity of BD [71]. In addition, the promoter of SLC6A4, encoding the serotonin transporter protein, has been found to hypermethylation in lymphoblastoid cell lines (LCLs) and postmortem brains of BD with lower mRNA expression only in BD carrying the S/S genotype [104].
Glutamate (Glu) is an excitatory neurotransmitter in the brain, which has a reciprocal effect with GABA which is an inhibitory neurotransmitter. Hyperactivation of the glutamatergic system [105,106] and hypofunction of the GABAergic interneuron appear to play important roles in the etiology of BD [107]. The SLC1A2 gene encodes the excitatory amino acid transporter (EAAT)-2, which transfers Glu from synapses to glial cells and has DMPs in its promoter region in BD with and without addiction using HRM-PCR, showing hypomethylation in addictive and hypermethylation in non-addictive [72]. In addition, in the postmortem brains of BD, reduced expression of glutamic acid decarboxylase1 (GAD1) results in decreased levels of GABA [73]. A group of genes including the Msh homeobox 1 gene, cyclin D2 gene, and death-associated protein 6 gene regulate GAD1 expression, which were termed as GAD1 regulatory network [73]. Diagnosis- and circuit-specific DNA methylation changes in several GAD1 regulatory network genes and their relationship with gene expression have been demonstrated in detail in the human hippocampus of BD, and these genes are mainly involved in chromatin regulation and cell cycle control [73]. Lastly, Reelin (RELN) can be expressed in GABAergic interneurons and glutamatergic neurons. There is no aberrant methylation in RELN was found in BD [94,108,109].
Due to the complex interconnections between central neurotransmitter systems, how abnormalities in central neurotransmitter function in BD are influenced by epigenetic mechanisms needs to be explored in more detail in the future. In addition, different episodes, different tissues, and disease stages should be considered.
DNA Methylation of Genes Associated with Immune System and InflammationInvolvement of the immune system has been proposed in the pathology of BD. Some main cytokines, including TNF-α, sTNF-R1, and sIL-2R, were found significant changes in BD [110]. As a negative critical controller of inflammatory response, suppressor of cytokine signaling-1 (SOCS-1) gene has been found higher DNA methylation levels in BD than in controls [78]. The human leukocyte antigen (HLA) complex group 9 gene (HCG9) has shown consistent hypomethylation in the brain, blood, and sperm of BD [111]. In 2016, Pal et al. have used new epigenetic technologies to rediscover hypomethylation of HCG9 in the postmortem prefrontal cortex of BD [79].
DNA Methylation of Neuroendocrine-Related Genes in BDThe dysfunction of the hypothalamic-pituitary-adrenal (HPA) system is suggested to underlie the pathophysiology of the onset and progression of BD, which may be associated with impaired stress resilience due to child trauma [112].
FK506 binding protein 5 (FKBP5) affects the negative feedback inhibition of the HPA system by regulating the sensitivity of glucocorticoid receptors (GRs). Hypermethylation of FKBP5 intron and elevated expression of this gene have been found in patients with BD [80]. Gene × environment interaction has been shown to contribute to BD pathogenesis by affecting DNA methylation levels in FKBP5 [80,81]. This mechanism of interaction has been found to be due to allele-specific methylation, in which populations carrying the risk allele may develop stress-related psychiatric disorders by changing DNA methylation status in FKBP5 when experiencing childhood trauma [113,114]. In addition, a study has found that the DNA methylation status of the glucocorticoid receptor1F (GR-1F) region was not associated with childhood trauma and was not altered in BD compared with controls [82].
It has been found that hypermethylation of the KIT ligand (KITLG) gene was associated with childhood trauma and mediated the lower cortisol stress reactivity resulting from childhood trauma [115]. However, hypomethylation of KITLG has been identified in BD but was not associated with childhood trauma [83]. The KITLG gene, known as stem cell factor, plays an important role in hematopoiesis, spermatogenesis, and melanogenesis. Therefore, hypomethylation of KITLG may affect the pathological mechanisms of BD by other molecular and biological mechanisms, which need to be further investigated.
DNA Methylation of Channel-Related Genes in BDVoltage-gated ion channels are a class of transmembrane proteins that play a major role in the regulation of neuronal excitability and responsiveness to synaptic inputs. A GWAS data in BD has suggested that four voltage-gated ion channel genes may contribute to the susceptibility of BD, including CACNA1E, KCNQ3, KCNMA1, and KCNIP4 (a regulatory subunit) [116]. CACNA1C is a protein coding gene and encodes calcium voltage-gated channel subunit alpha1. Hypermethylation has been found in CACNA1C in peripheral tissues of BD, which is influenced by SNPs within the intron 3 risk locus of CACNA1C [84]. KCNQ3 is a member of a family of voltage-gated potassium channel genes and its coded protein is active in neurons contributing to signal transmission in the brain. A study has demonstrated that DNA methylation levels of CpGs located in exon 11 of KCNQ3 gene were significantly lower in BD patients’ postmortem prefrontal cortex, which were significantly positively correlated with mRNA levels [85].
In summary, these studies indicated that abnormal DNA methylation status of voltage-gated ion channel genes may result in dysregulated ion channel function. This potentially plays an important role in the pathogenesis of BD. The exact pathogenic mechanism remains unclear and further research is needed.
DNA Methylation of Circadian Rhythm Genes in BDThe circadian rhythm is a system that organizes a range of biological processes in 24 hours, from sleep to neural activity [117]. It has been observed that BD patients have higher rates of interruption of biological rhythm compared with healthy controls, after controlling the influence of other variables such as sex, alcohol, tobacco, other psychiatric disorders, and psychotropic medication use [118]. Genetic changes in circadian genes may destroy normal circadian rhythm and increase the risk of BD developing [118].
There are a few circadian rhythm genes related to BD that have been studied, such as aryl hydrocarbon receptor nuclear translocator-like (ARNTL) gene, and melatonin receptor type 1A (MTNR1A). ARNTL activates the promoter E-boxes of monoamine oxidase A (MAOA) gene and regulates the gene expression of MAOA as a heterodimer with NAPS2 [119]. Hypermethylation at cg05733463 in ARNTL has been identified in BD [86]. Melatonin secretion alters the circadian rhythm and sleep-wake cycle, and MTNR1A is the melatonin receptor A gene. In 2023, Lesicka et al. have found that the methylation level of MTNR1A was decreased in depressed patients (with unipolar and bipolar disorders) compared with healthy controls [87].
There was a lack of literature on DNA methylation abnormalities of other circadian rhythm genes in BD, such as CLOCK and PER3, to the best of our knowledge. However, these genes have been suggested to be associated with BD etiology [120,121].
Candidate Genes from Genetic Studies in BDDNA methylation of several other candidate genes from genetic studies of BD has been studied, such as DTNBP1 and FAM63B. DTNBP1 plays an important role in the function of neurons by influencing glutamate release via upregulation of pre-synaptic machinery and is a susceptibility gene for SCZ [122]. The higher methylation of CpG sites in the upstream of DTNBP1 gene has been found in female BD patients compared with healthy controls [94]. Abdolmaleky et al. have also shown that BD patients with psychotic depression exhibited hypermethylation of DTNBP1 versus other BD patients, suggesting that it could be a marker of psychotic phenotype [88].
A methylome-wide association study (MWAS) in SCZ patients noticed many top sites, and one of them was in FAM63B gene [123]. Due to the high degree of comorbidity between SCZ and BD, Starnawska et al. assessed the methylation status of two CpG sites of FAM63B in whole blood samples from BD patients and found a lower methylation of FAM63B [89].
BDNF is a neurotrophic growth factor family, which plays an important role in synaptic transmission, neurogenesis, and cognitive function. The DNA methylation status of BDNF has been widely studied in many aspects including different episodes [74,124], pharmacotherapy [74,124], and cohort studies [75]. Converging evidence has demonstrated that significantly higher methylation levels of the BDNF promoter were found in MDD and BDII, but not in BDI [74,75,124]. Patients on pharmacological treatment with Li and valproate have shown decreased DNA methylation levels of BDNF promoter, and antidepressant treatment increased DNA methylation levels of BDNF promoter [74,76,124]. The DNA methylation levels of BDNF in brain tissue should be studied in the future, as it can serve as a proxy for brain-specific alterations [77].
Methylation-related bioinformatic approaches have taken off and it has been of course used to study BD. Recently, more and more functional genomics data, including epigenetic data, can be integrally analyzed to understand epigenetic variation and its impact. This is an area worthy of further study in the future. The methylation-related multi-omics data analysis studies on BD were summarized in Table 5.
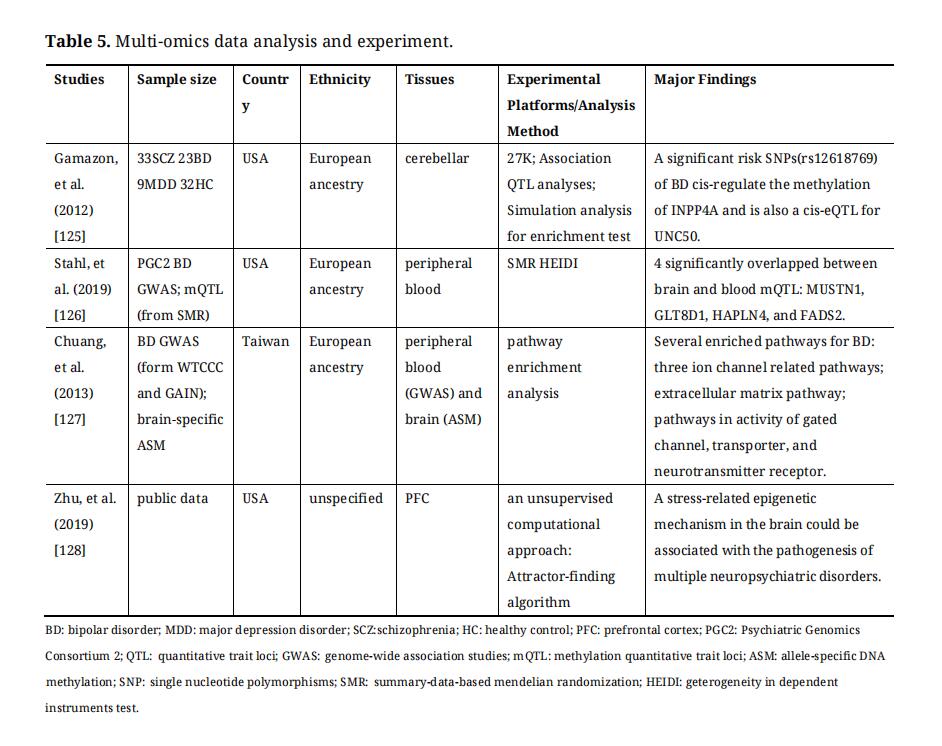 Table 5. Multi-omics data analysis and experiment.
Table 5. Multi-omics data analysis and experiment.
In addition to the environment, genetic variation can also influence epigenetic modifications. As genetic–epigenetic interactions, mQTL (methylation quantitative trait locus) and ASM (allele-specific DNA methylation) can be used in finding DNA methylation abnormalities in BD by integrating with GWAS data of BD [129]. A part of DMRs from the profiling of hap-ASM (haplotype-dependent ASM) and mQTLs are specific for different types of cells, and the DMRs always were located near GWAS signals for immune and neurological disorders [130]. Gamazon et al. have analyzed mQTL combined with eQTL (expression quantitative trait locus) data which provided ways of functionally annotating genetic variation from GWAS for the pathophysiology of BD, and found a risk SNP (rs12618769) which cis-regulate the methylation of INPP4A and is also a cis-eQTL for UNC50 [125]. The protein encoded by INPP4A gene is a target for the effects of lithium that can prevent excitatory neuronal cell death and maintain the functional integrity of the brain [125]. SMR (Summary data-based mendelian randomization) and HEIDI (heterogeneity in dependent instruments) tools have been developed to analyze the correlation between DNA methylation or gene expression and phenotype, using summary-level SNP data from independent GWAS, mQTL, and eQTL studies [131]. Using SMR and HEIDI tests, four CpGs have been identified which were associated with BD both in the brain and blood, and the four mQTLs were located near MUSTN1, GLT8D1, HAPLN4 and FADS2 [126].
ASM describes the phenomenon that the statuses of DNA methylation are affected by genetic polymorphisms in a cell. This type of DNA methylation site is distributed in the whole genome. Li-Chung Chuang et al. have integrated a brain ASM dataset and two GWAS datasets of BD and then identified many of the biological pathways that weren’t significant in GO analysis of BD GWAS, including gated channel, transporter, and neurotransmitter receptor [127].
Furthermore, due to the extensive linkage disequilibrium (LD) of the genome, identifying causal genetic variations by mQTL is not very reliable. Therefore, a new deep learning model, INTERACT, has been developed to predict the effects of genetic variations on DNA methylation levels in the human brain by integrating convolutional neural networks with transformer [132]. The genetic variations indicated by INTERACT are not confounded by LD. In patients with schizophrenia, this model could improve polygenic risk prediction across diverse ancestry samples and can be used to identify the functional regulatory variants through integrated analysis with GWAS risk loci [132].
All these analytical methods can be used to study epigenetic mechanisms underlying the pathogenesis of BD. In addition, researchers should develop and use new methods to integrate multi-omics data to better explain the pathogenesis of BD.
Co-Methylation Network AnalysisIn the genome, a set of DNA methylation positions or regions might form a module in which their methylation levels might simultaneously change, causing specific biological processes. MWAS is only able to detect individual DMPs and DMRs, and these independent DMPs and DMRs limit the abilities to explain biological processes from the structural and functional complexity of the genome and the dynamic nature of epigenetic modification. On the other hand, due to multiple testing corrections, many CpGs with important biological functions may be missed in MWAS. Now, co-methylation network analysis can be used in DNA methylation studies because of its ability to identify co-methylation network modules and increase statistical power. Furthermore, co-methylation, co-expression, and protein-protein interactions can be used for gene-module detection in multi-omics datasets [128,133]. A meta-analysis has integrated expression and methylation profiles in the brains of patients with multiple neuropsychiatric diseases (including SCZ, BD, etc.), and has found the pathogenesis of these diseases was all associated with a stress-related epigenetic mechanism [128].
Site-specific DNA methylation detection, MWAS, and integrative analysis of multi-omics data can find some significant DMPs, DMRs, and biological pathways in BD. Based on the above results, it is important to explore the role of DMPs and DMRs in biological processes contributing to the pathogenesis of BD by conducting wet lab experiments, such as dual-luciferase reporter assay systems and DNA methylation editing methods.
The dual luciferase assay has been widely used in cell lines to determine rapidly and accurately the activity of a given promoter and/or enhancer. The CpG-Free plasmids contain elements that include genes encoding selectable markers, reporters, and others, and they are artificially lacking CpG dinucleotides. Because of this feature, there is no concern that DNA methylation will appear on the plasmid backbone and affect reporter gene expression. This kind of plasmids can be used in to dual-luciferase reporter assay system and explore the impact of DNA methylation of the promoter and enhancer region on transcriptional activity in vitro, regardless of the effect of CpG DNA methylation on its backbone [134]. Cortical GAD1 mRNA levels have been reported to be reduced in BD. Using a dual-luciferase reporter gene experiment, Chen et al. have found that a decrease in the level of DNA methylation in the 5' untranslated region of GAD1 was accompanied by an increase in GAD1 expression, mediated by cis-regulatory elements and VPA-induced dose-dependent histone acetylation [135]. There is a limit to this kind of experiment: it cannot directly represent the effect of DNA methylation-related functional elements on gene expression but reflects the effect indirectly by the expression of the reporter gene. Therefore, more exact experiments need to be exploited for DNA methylation studies in BD.
Many targeted DNA methylation and demethylation methods have been developed by coupling targeted DNA recognition platforms with a methylation writer or eraser protein. In 2016, Liu et al. established a DNA methylation editing method based on CRISPR gene editing technology to achieve targeted DNA methylation editing through the fusion of Tet1 or Dnmt3a with catalytically inactive Cas9 (dCas9) [136]. This study showed that targeting the dCas9-Tet1 fusion protein to methylated BDNF promoter IV and MyoD distal enhancer can induce demethylation and activate the two genes' expression [136]. In addition, the authors have further demonstrated that by using dCas9-Dnmt3a in mice, the targeted de novo methylation of CTCF-binding sites can be induced, and then local DNA looping and gene transcription in the neighboring loop were altered [136]. According to previous studies, abnormal DNA methylation can lead to altered expression of key genes associated with disease development. Using dCas9-Tet1 or -Dnmt3a for targeted DNA methylation editing in hiPSCs (human induced pluripotent stem cells), the epigenetic-based therapeutic approaches of diseases have been explored, including FXS (Fragile X syndrome) [137] and PD (Parkinson’s disease) [138]. DNA methylation editing technologies are constantly evolving and an increasing number of new tools are being developed [139]. These technologies can provide new insights into how DNA methylation regulates gene expression. Therefore, it is possible to clarify how DNA methylation affects disease development through gene expression and explore the epigenetic-based therapeutic approach to diseases. DNA methylation editing technologies are yet to be used in DNA methylation studies in BD. It is promising to explore the pathogenesis and epigenetic treatment of BD by using these technologies.
DNA methylation is stable, inherited, and reversible, and it may provide a possibility to understand the etiology of BD. Moreover, it is a promising biomarker for the diagnosis and response to disease treatment of BD. Recently, the DNA methylation studies in BD are still lacking. A lot of studies with a candidate gene strategy have demonstrated that changes in DNA methylation of many genes are associated with BD, but changes in DNA methylation underlying the pathophysiological basis of BD need further clarification. MWAS also found a few meaningful genes and pathways of methylomic differences in BD. However, due to its high cost, large-sample MWAS of BD was difficult to conduct. Therefore, multi-omics analysis based on microarrays and next-generation sequencing technologies may provide an effectivity method to cover the recent shortage for discovery and validation of changes in DNA methylation in BD. In addition, there is a mount of public data online, including many BD GWAS and mQTL datasets, and biological information methods, such as SMR, can be used to identify novel DNA methylation sites and explore their underlying biological mechanisms.
DNA methylation editing technologies provide more effective tools to precisely manipulate the DNA methylation status of specific genomic regions. This technology can be used to clarify how DNA methylation affects gene transcription in vivo or in vitro [136]. Using multi-omics data analysis combined with DNA methylation editing experiments is expected to explore the mechanism of DNA methylation underlying the pathogenesis of BD further and systematically.
No data were generated from the study.
All authors declared that there is no conflict of interest exists in this study.
This research was supported by grants from the National Research and Development Grant from the Ministry of Science and Technology (Grant No. 2021YFE0191400 (X.C.)), and National Natural Science Foundation of China (Grant No. 82101576 (Z.L.), 81871056 (X.C.)). No funding sources had roles in study design, data collection, analysis and interpretation of data, decision to publish or preparation of the manuscript.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
80.
81.
82.
83.
84.
85.
86.
87.
88.
89.
90.
91.
92.
93.
94.
95.
96.
97.
98.
99.
100.
101.
102.
103.
104.
105.
106.
107.
108.
109.
110.
111.
112.
113.
114.
115.
116.
117.
118.
119.
120.
121.
122.
123.
124.
125.
126.
127.
128.
129.
130.
131.
132.
133.
134.
135.
136.
137.
138.
139.
Yang Z, Zhang S, Ouyang L, Liao A, He Y, Li Z, et al. DNA Methylation and Bipolar Disorder. J Psychiatry Brain Sci. 2023;8:e230012. https://doi.org/10.20900/jpbs.20230012

Copyright © 2023 Hapres Co., Ltd. Privacy Policy | Terms and Conditions




